
Genes for sexual body size dimorphism in hybrid tilapia (Oreochromis sp. x Oreochromis mossambicus)
2019-12-05 01:49ZiYiWanGraceLinGenhuaYue
Aquaculture and Fisheries 2019年6期
Zi Yi Wan, Grace Lin, Genhua Yue,c,*
a Molecular Population Genetics and Breeding Group, Temasek Life Sciences Laboratory, 1 Research Link, National University of Singapore, 117604, Singapore
b School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, 637551, Singapore
c Department of Biological Sciences, National University of Singapore, 14 Science Drive 4, 117543, Singapore
Keywords:Sexual dimorphism Body size RNA-Seq Tilapia Aquaculture Evolution
A B S T R A C T Tilapias are important aquaculture species. Male-biased sexual size dimorphism is very common and males are preferred for aquaculture in tilapia. However, the mechanisms underlying sexual dimorphism remain to be elucidated. One hundred and thirty-six sex-biased genes, of which 23 were male-biased and 113 were femalebiased, were identified via reanalysis of a muscle transcriptome data using the latest reference genome assembly.These genes were mapped to KEGG pathways that are related to somatic cell metabolism, growth and differentiations, such as MAPK, FoxO4 and metabolism pathways as well as developmental processes responsible for skeletal muscle development. Pathways related to cell growth and proliferations such as MAPK signaling pathways are upregulated in males while pathways regulating cell division such as FoxO4 are upregulated in females. Sex-biased genes in tilapia skeletal muscle have higher evolution rates (dN/dS) compared to unbiased genes. Female-biased and male-biased genes showed 17.4% and 13.5% higher dN/dS, respectively, compared to unbiased genes. Our results suggest that some of the male and female sex-biased genes were under selection pressures. Three SNPs located in the promoter region of one sex-biased gene RASGRF1 on LG1 were associated with bodyweight differences in the hybrid tilapia. These sex-biased genes identified in this study may serve as candidate genes for future functional analysis on sexual size dimorphism and for developing DNA markers for selecting fast-growing saline tilapia in aquaculture.
1. Introduction
Sexual dimorphism refers to the marked phenotypic difference between males and females from the same species (Chervinski, 1965;Fairbairn, Blanckenhorn, & Székely, 2007; Isles, 2002). These differences may be in terms of morphology, physiology, gene expressions or behaviors (Ellegren & Parsch, 2007; Fairbairn et al., 2007; Parsch &Ellegren, 2013). For male-biased sexual size dimorphisms, larger males may be favoured through both natural selection and sexual selection(Fairbairn et al., 2007; Parsch & Ellegren, 2013; Ralls & Mesnick, 2009).Through natural selection, larger males are at the advantage of producing more milt with increasing body size, hence allowing a higher rate of fertilization success in a polygynandry breeding set. Through sexual selection, larger males with faster growth rate may have the advantage on male-to-male combat during breeding seasons and have a higher chance of mating with females on the breeding grounds. Under a no environmental constraint model, larger males may have access to more mates and allow for a higher number of spawning than smaller males (Parker, 1992). It is believed that the degree of sexual dimorphism is the result of the difference, between males and females, in terms of the sum of all the selective pressures (natural selection and sexual selection) affecting them (Fairbairn et al., 2007; Parker, 1992;Ralls & Mesnick, 2009). The consequences of sexual dimorphism on the ecology and evolution of organisms are often profound (Ellegren &Parsch, 2007; Fairbairn et al., 2007; Isles, 2002; Parsch & Ellegren,2013). Therefore, it is important to understand molecular mechanisms underlying sexual dimorphism in a species.
On Earth, there are over 30,000 species of teleosts (Nelson, Grande,& Wilson, 2016). In some fish species, including tilapia andCynoglossus semilaevis, sexual dimorphisms are very obvious and diverse. For example, tilapia males have a faster growth rate than females while the opposite phenotype can be observed inC. semilaevis(Chervinski, 1965;Wang, Wang, Wang, & Chen, 2018; Wang et al., 2016). However, in some species such as zebrafish, there is no major phenotypic difference between the sexes. Sexual size dimorphism in fish are of importance in aquaculture. Uneven growth rate between the sexes often leads to uneven harvest size, poor feed conversion efficiency and stunted growth in the sex with a slower growth rate (Lind et al., 2015).Therefore, understanding more about the mechanisms underlying sexual size dimorphism will enable researchers to develop technologies for producing the preferred sex for aquaculture production.
Tilapia is the common name of Nile tilapia (Oreochromis niloticus),Mozambique tilapia (Oreochromis mossambicus), Blue tilapia(Oreochromis aureus) and other cichlid species (Nelson et al., 2016).Amongst all tilapia species,O. niloticusis the most important species for aquaculture (Ye et al., 2006). In particular, O. niloticusis the most important tilapia species for freshwater aquaculture, whileO. mossambicusis able to adapt to full salinity seawater. However,O. mossambicusgrows at a much slower rate thanO. niloticus, diminishing its potential as an aquaculture species. In the past years, various groups have tried to crossO. mossambicusandO. niloticusto generate a hybrid tilapia, which can grow faster thanO. mossambicusand adapt to full seawater (Wang, Cui, Yang, & Cai, 2000; Watanabe, Kuo, & Huang,1985). Many in the tilapia farming sector worldwide have been producing all-male fingerlings to raise yield and prevent reproductions(Lind et al., 2015). These sex-reversed populations relied on hormone treatments during early fingerling stage to induce masculinization,while some experimental phase tilapia YY supermale have been developed to produce full XY male populations for tilapia aquaculture (El-Greisy & El-Gamal, 2012; Lind et al., 2015; Scott, Penman, Beardmore,& Skibinski, 1989). However, our current understanding on the origin and mechanisms of sexual size dimorphism in tilapia and other aquaculture species is still in its infancy (Shen & Yue, 2019).
So far, it is understood that gonads are responsible for regulating growth in Mozambique tilapia, as demonstrated by Bhatta et al. inO.mossambicus(Bhatta et al., 2012). Complete gonad removal via gonadectomy significantly retarded growth compared to control fishes (with sham gonadectomy). Ectopic transplantation of gonadal tissue reversed the growth retardation caused by gonadectomy. That study provided evidence that gonads are integral players in regulating growth inO.mossambicus. However, how sexual organs (e.g. testis and ovary) of different sex lead to different growth rates are not well understood.Interestingly, QTL studies on tilapia have shown that male and female tilapia have different QTL profiles for traits such as body weight and total length. These results suggest that genetic factors may be regulated differentially in each sex, resulting in different growth rates in male and female tilapia (Lin, Chua, Orban, & Yue, 2016; Liu et al., 2014). In our previous study, we showed that genome-wide methylation analysis identified sexually dimorphic methylated regions in hybrid tilapia (Wan et al., 2016), while which genes and regulatory elements are involved in sexual dimorphism remains largely unknown. Analysis of transcriptomes is an important way to identifying differentially expressed genes related to important traits as demonstrated in previous studies(Batista, Churcher, Manchado, Leitão, & Power, 2019; Liu, Xia, Pang, &Yue, 2017; Qian, Ba, Zhuang, & Zhong, 2014; Xiao et al., 2019).
The purpose of this study was to identify genes for sexual size dimorphism to understand more about the molecular mechanisms underlying it and the evolution of sex-biased genes in saline hybrid tilapia,as well as to detect SNPs for selecting fast-growing hybrid tilapia for aquaculture in brackish water or full seawater (i.e. salinity 30-34 ppt).We reanalyzed the muscle transcriptome data used in the analysis of genome-wide methylation (Wan et al., 2016) using the latest reference genome O_niloticus_UMD_NMBU, which includes resolved complex regions that were previously unavailable in the older version of the reference genome (Orenil1.1). We detected 136 sex-biased genes and showed that some sex-biased genes were evolving at a faster rate than unbiased genes. In addition, we identified SNPs associated with bodyweight in the promotor region of one (RASGRF1) of the sex-biased genes. These SNPs may be used for selection of fast-growing tilapia.
2. Materials and methods
2.1. Fish, sampling, RNA extraction and RNA sequencing
The tilapia population used in this study was from an F2hybrid tilapia line derived from crossing Mozambique tilapia and Nile tilapia.The hybrid tilapia could grow out in a full seawater (30-34 ppt). The culture of the hybrid F2tilapia was described in detail in a previous paper (Wan et al., 2016). Muscle samples were collected from four tilapias, including Male 1 (M1), Male 2 (M2), Female 1 (F1) and Female 2(F2) at the age 108 days post hatch, and were stored in TriZOL (Thermo Fisher, Waltham, USA) for RNA extraction. These hybrid tilapia shows male-biased sexual size dimorphisms. RNA extraction from the muscle samples, the construction of libraries and RNA sequencing on a Illumina HiSeq 2000 were described in a previous paper (Wan et al., 2016).
2.2. Identification of sexually dimorphic differentially expressed genes(DEGs)
Transcriptome data generated in a previous study (Wan et al., 2016)were downloaded from NCBI repository under BioProject number PRJNA293932 using SRA toolkit command line version. Reads were filtered and checked for quality control using the IlluQC.pl module from NGSToolKit Version 2.3 (Patel & Jain, 2012). Low quality reads were discarded. Only reads that passed IlluQC.pl default settings were retained. The Nile tilapia reference genome used was downloaded from GenBank at the accession number GCA_001858045.3. The reference genome was then indexed for STAR alignment using the command STAR -runMode genomeGenerate. Good quality reads were then mapped onto the Nile tilapia reference genome using the software STAR(Dobin et al., 2013). The alignment BAM files were then loaded onto SeqMonk to calculate the log2Reads per Million Sequenced Read (RPM)using the mRNA annotation track (.gtf file) from NCBI database corresponding to O_niloticus_UMD_NMBU assembly (Conte,Gammerdinger, Bartie, Penman, & Kocher, 2017). Datasets were normalised to remove biasness from read coverage as well as PCR duplicates using SeqMonk (https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/). Read counts were then calculated for each gene using the software HTSeq-count (Anders, Pyl, & Huber, 2015). Read counts were then merged and loaded into the R-environment for DESeq2 differential gene expression analysis (Love, Huber, & Anders,2014) to detect sex-biased genes. We applied a cut-offthreshold of false positive corrected p-value (p-adj) of 0.05, with a gene expression foldchange of at least two to define a gene as being sex-biased. To understand the overrepresentation of the DEGs in our transcriptome analysis,we extracted the mRNA FASTA sequences of all DEGs from NCBI.FASTA sequences were BLAST searched againstDanio rerioEnsembl cDNA FASTA database with TBLASTN command line version, specifyingDanio reriogene only. BLAST top hits with e-values satisfying the cut-offpoint of lower than 1e-4were retained and corresponding zebrafish orthologue for each gene was assigned ( Altschul et al., 1990).We uploaded these 126 Ensembl IDs into PANTHER DB (http://www.pantherdb.org/geneListAnalysis.do) for statistical overrepresentation test (Mi et al., 2010). We applied Fisher's exact test, with false discovery rate (FDR) calculation with a cut-offof 0.05. Gene ontology IDs were extracted from Ensembl BioMart and analysed on PANTHER and KEGG for enrichment analysis (Kanehisa & Goto, 2000; Mi et al., 2010).
2.3. qRT-PCR for the validation of the expressions of DEGs identified with RNA-seq
Four male and four female tilapia fish at 108 dph were euthanized in AQUI-S (AQUI-S, Lower Hutt, New Zealand) at overdose condition.Skeletal muscle tissue was collected and snap frozen in liquid nitrogen until ready for RNA extraction. RNA extraction was carried out using TRIzol (Invitrogen, Carlsbad, USA) according to the manufacturer's protocol and eluted in RNase-free water. Reverse transcription was carried out using M-MLV reverse transcriptase (Promega, Fitchburg,USA) with a mix of random hexamer primers and oligo-dT primers.Primers were designed from six sex-biased genes to span exon-exon junctions with product size ranging from 220 bp to 300 bp, and with annealing temperature of 58 °C. Real time PCR was conducted using KAPA SYBR Fast Universal Kit (Sigma-Aldrich, Missouri, United States)in CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad Laboratories, California, United States). qPCR data analysis was conducted using the delta delta Ct method. Primer sequences for qPCR analysis can be found in Supplementary Table 1.
2.4. Identifying SNPs in a sex-biased gene RASGRF1 and analyzing their associations with bodyweight
To identify possible SNPs close to candidate sex-biased genes that may be associated with body weight, we designed a pair of primers located on the promoter region ofRASGRF1. Primers were designed with Primer3 software hosted on the web (http://bioinfo.ut.ee/primer3/) with annealing temperature target of 55 °C. Primer sequence can be found in Supplementary Table 1. One hundred and fiftyeight samples from a family, which were previously used to conduct QTL mapping for growth traits in saline tilapia (Lin et al., 2016), was used for genotyping SNPs to examine their associations with growth.Fish were raised in full saline water up to 220 dph. At 220 dph, fish were anaesthetized with AQUI-S (AQUI-S, Lower Hutt, New Zealand)and weighed on a digital scale (Mettler Toledo, Ohio, United States).Fin clips were sampled and stored in absolute ethanol at -20 °C until DNA extraction. DNA were extracted from 158 samples of tilapia fin clips using a method described by Yue and Orban (Yue and Orban,2005). PCR was done using the following thermal cycling condition:3 min at 95 °C followed by 40 cycles of 30 s at 95 °C, 30 s at 55 °C and 1 min at 72 °C and final step at 72 °C for 10 min. Genotyping was performed by direct sequencing of PCR products with forward primers and BigDye chemicals on an ABI 3730xl (Thermo Fisher Scientific, Massachusetts, United States). SNPs calling were performed using Sequencher V5.1 (Gene Codes Corporation, Michigan, United States). Association between body weight and SNPs were calculated with ANNOVA in Microsoft Excel 2013.
2.5. Calculation of synonymous and nonsynonymous mutation rates, and codon adaptation index
To estimate the evolution rate of sex-biased genes identified in this study, we downloaded coding sequences and protein sequences of closely related cichlid species from Ensembl, includingMaylandia zebra,Amphilophus citrinellus, Neolamprologus brichardi, Astatotilapia burtoniandHaplochromis nyererei(Brawand et al., 2014; Zerbino et al., 2018).All protein-coding sex-biased genes were used to calculate nonsynonymous and synonymous mutation rates (dN/dS). Thirty-six random non-biased genes were selected for synonymous and nonsynonymous mutation rate calculations, to represent the non-biased gene groups.Alignment of protein sequences was performed with ClustalOmega using default parameters (Sievers et al., 2011). Codon alignment of coding sequences of orthologues and Nile tilapia sequence was performed with PAL2NAL guided by protein FASTA alignments generated by ClustalOmega (Suyama, Torrents, & Bork, 2006). Only the first gapless alignment segments were selected fordN/dScalculations. Synonymous and nonsynonymous mutation rate calculations were performed with the codeml software from the PAML package (Yang, 2007).PairwisedN/dScalculations were performed with F3X4 codon frequency model in pairwise mode (runmode =-2). The codeml output was parsed with a python script parse_codeml_output.py (https://github.com/faylward/dnds). Statistical tests comparingdN/dSdifferences between gene groups were done with Mann-WhitneyUTest using Microsoft Excel 2013.
To calculate the codon adaptation index (CAI), coding sequences of sex-biased genes and non-biased genes for tilapia were downloaded from BioMart and combined into three multi-fasta files representing male-biased, female-biased and unbiased genes. Coding sequences were then uploaded onto the E-CAI server (http://genomes.urv.es/CAIcal/ECAI/) for CAI and expected CAI (eCAI) calculations (Puigbò, Bravo, &Garcia-Vallvé, 2008). The Codon Usage Reference Table used for CAI and eCAI calculations was downloaded from the Codon Usage Database(http://www.kazusa.or.jp/codon/) (Nakamura, Gojobori, & Ikemura,2000). Statistical tests to compare the CAI differences between groups were conducted with Mann-WhitneyUTest in Microsoft Excel (2013).
3. Results
3.1. Identification of sexually dimorphic differentially expressed genes(DEGs)
RNA-seq libraries were sequenced on a HiSeq 2000, generating a total of 15.4 GB of data with 76.5 million paired-end reads. On average,each library was made up of 19.1 million paired-end reads. The Nile tilapia reference genome assembly used in this RNA-seq analysis is the O_niloticus_UMD_NMBU assembly. The percentage of uniquely mapped reads for M1, M2, F1 and F2 were 92.33%, 92.93%, 92.91% and 92.87% respectively. The number of reads and mapping efficiency of each library were summarized in Supplementary Table 2. Differentially expressed genes (DEGs) were defined as having a log2fold-change of more than 1/-1 and an adjustedp-valueof less than 0.05. In our analysis, we found 136 sexually dimorphic DEGs in the skeletal muscle transcriptome. Twenty-three genes were highly expressed in the male tilapia transcriptome, while 113 genes were highly expressed in the female tilapia transcriptome. Among the 23 male-biased genes, two genes belong to the non-coding RNA class while the rest are coding RNA. Out of the female-biased genes, four genes belong to the noncoding RNA class while the remaining genes are protein coding RNAs.The five genesRASGRF1,LPIN1,FOXO4,IRS2andEGFR, which are known to be related to somatic growth, were in the list of the 136 sexbiased genes. The non-coding RNAs identified to be sex-biased in expressions are LOC109195677, LOC102079801, LOC106099004,LOC112841878, LOC112841790 and LOC112843301. These are novel non-coding RNAs and their functions are unknown. A survey of these six genes on BLAST revealed no similar orthologues reported in model organisms. A summary of all sex-biased genes identified is listed in Supplementary Table 3.
We assigned 126 zebrafish orthologues to 136 of all sex-biased genes identified. Ten genes showed no orthologues from zebrafish genomes, including the six novel non-coding RNAs, while four others had no orthologues. From the 126 orthologues, we observed overrepresentation in genes relating to some biological processes including muscle system process (GO:0003012), anatomical structure morphogenesis (GO:0009653), actin filament-based process (GO:0030029),multicellular organismal process (GO:0032501), actin cytoskeleton organization (GO:0030036), regulation of muscle contraction(GO:0006937), regulation of muscle system process (GO:0090257),circadian regulation of gene expression (GO:0032922) and muscle contraction (GO:0006936). On the cellular compartment GO segment,troponin complex (GO:0005861), myofilament (GO:0036379), striated muscle thin filament (GO:0005865), sarcomere (GO:0030017), contractile fiber part (GO:0044449), myofibril (GO:0030016), contractile fiber (GO:0043292), I band (GO:0031674), actin cytoskeleton(GO:0015629), supramolecular fiber (GO:0099512), supramolecular polymer (GO:0099081), supramolecular complex (GO:0099080) and cytoskeleton (GO:0005856) were overrepresented (Fisher's Exact test,P<0.05).
Using db2db server conversion, we obtained KEGG object identification for each zebrafish orthologue. We then mapped these KEGG IDs using KEGG Mapper (https://www.kegg.jp/kegg/tool/map_pathway1.html) and searched against the dre (Danio rerio) database(Kanehisa &Goto, 2000). The pathways with the most hits are the metabolism pathway (dre01100),MAPKsignaling pathway (dre04010) andFoxOsignaling pathway (dre04068) with eight, five and three gene hits respectively. A summary of all KEGG pathways mapped is listed in Table 1.

Table 1 Sex-biased genes and KEGG pathways mapped by using Danio rerio orthologues of sex-biased genes in hybrid tilapia (Oreochromis sp. x Oreochromis mossambicus). Only pathways with more three and more genes mapped to the KEGG Danio rerio (dre) pathway database are shown.
3.2. Validation of RNA-seq data with qRT-PCR
To validate our RNA-seq data, we selected six genes from sex-biased genes identified, and designed primers on the 3’ end of the mRNA sequences (Supplementary Table 1.). Using theactingene as a normalization control, we analyzed the expression data using the delta-delta Ct method. Using a regression analysis of RNA-seq log2(RPM) values and actin-normalized expression values, we obtained aR2value of 0.951 and 0.821 for male and female DEGs analyzed, respectively. Thepvalue for the regression analysis werep= 0.001 andp= 0.012, respectively, for male and female sex-biased genes.
3.3. Genomic locations of DEGs
Based on the latest genome assembly, O_niloticus_UMD_NMBU, we performed a Chi-square analysis to test if the differentially expressed genes are randomly distributed across various linkage groups in the tilapia genome. If the genes are randomly distributed across the linkage groups, the number of sex-biased genes in each linkage group should be proportional to the number of genes within the chromosome. By comparing the observed and expected numbers of sex-biased genes for each chromosome, we rejected the null hypothesis that the sex-biased genes are randomly distributed across the genome (Chi-squared testP<0.001). After calculating the Z-score of sex-biased gene count for each chromosome, we found that chromosomes LG4 and LG18 have Zscores of more than 1.645 (significant atP<0.05), suggesting that sex-biased gene expressions in the muscle tissues are enriched in these chromosomes as shown by Fig. 1. Three genes were also found to be within a QTL for growth previously identified in tilapia in chromosome LG18 (Lin et al., 2016). These genes are LOC100706127, CADM3 and a non-coding RNA (LOC109195677). The functions of LOC100706127 are unknown. A protein BLAST search showed the presence of an immunoglobin domain and similarity with proteins from signalling lymphocytic activation molecule (SLAM) family, suggesting a role in immune functions. CADM3 function as a cell to cell adhesion protein. The expression of CADM3, also known as NECL1, can suppress the growth and tumorigenic ability of colon cancer cells (Raveh, Gavert, Spiegel, &Ben-Ze'ev, 2009). The role of CADM3 in skeletal muscle growth is still unknown. The non-coding RNA LOC109195677 is a novel non-coding RNA with no known orthologues in other species.
3.4. Identification of SNPs in RASGRF1 promoter region and their association with bodyweight in hybrid tilapia
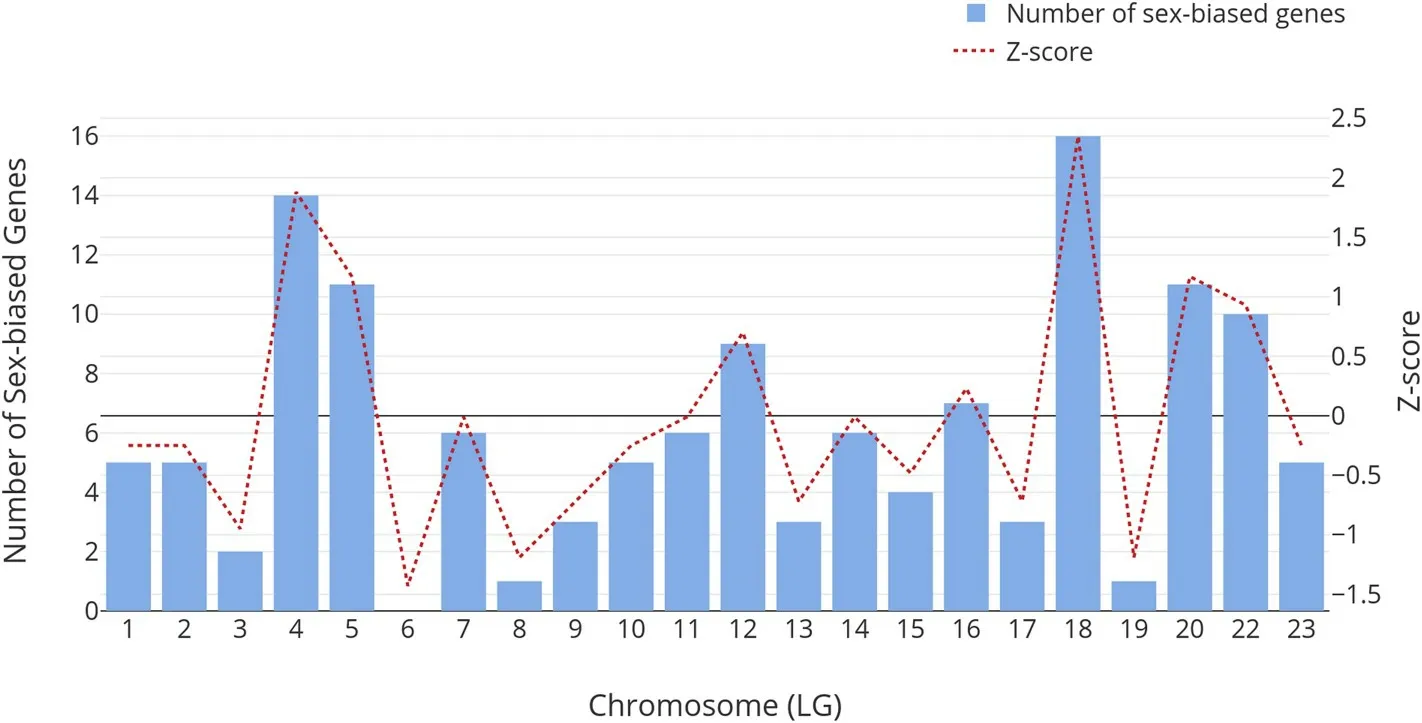
Fig. 1. Numbers of sex-biased genes on different chromosomes are shown on the bar plot. Z-scores were calculated and shown on the right Y-axis with the line graph.Chi-square analysis showed that the sex-biased genes are not randomly distributed across the genome (P <0.001).
To genotype and discover potential markers that may be applied in marker assisted selection for fast growth, we looked for SNPs close to the proximity of candidate genes. We identified three SNPs located at the promoter region ofRASGRF1. The coordinates of these SNPs are LG1:28146922 (CT/TT), LG1:28146925 (CT/CC) and LG1:28146966(AG/GG), based on the genome assembly O_niloticus_UMD_NMBU. The location of the SNPs LG1:28146922 (CT/TT), LG1:28146925 (CT/CC)and LG1:28146966 (AG/GG) are at 877 bp, 880 bp and 921bp upstream of the transcriptional start site of RASGRF1 respectively. In the paternal tilapia, the alleles in three SNPs are LG1:28146922 (TT),LG1:28146925 (CC) and LG1:28146966 (GG). In the maternal tilapia,the alleles in three SNPs are LG1:28146922 (CT), LG1:28146925 (CT)and LG1:28146966 (AG). In our genotyping population of 158 samples,we did not find any recombination between these three SNPs. From our population, individuals with the genotypes LG1:28146922 (CT),LG1:28146925 (CT) and LG1:28146966 (AG) weigh on average 350.9 g(n = 122) while individuals with the genotypes LG1:28146922 (TT),LG1:28146925 (CC) and LG1:28146966 (GG) weigh on average 315.7 g(n = 36). ANNOVA tests between these three SNP markers and bodyweight at 220 dph showed significant difference between fast growing and slow growing fish at 220 dph atP<0.05. Summary of the SNPs identified are described in Supplementary Table 4.
3.5. Female sex-biased genes exhibit higher evolution rates
Some sex-biased genes in tilapia skeletal muscles are evolving at a faster rate than non-biased genes (Mann-WhitneyUTest;P<0.001),having higherdN/dSratio as shown in Fig. 2. When we compared female-biased genes against non-biased genes, we observed significantly higher nonsynonymous to synonymous ratio (dN/dS) in female-biased genes (Mann-WhitneyUTest;P<0.001). However, when we compared male-biased gene against non-biased genes, we observe no significant differences (Mann-WhitneyUTest;P= 0.138). We also compared the mediandN/dSvalues for female-biased and male-biased genes against unbiased genes and found that they are 17.4% and 13.5%higher than unbiased genes, respectively.dN/dSof all sex-biased genes and unbiased genes are shown in supplementary materials(Supplementary. Tables 5-7).
We found significantdNdifference between female-biased and unbiased genes at 0.0052 (Mann-WhitneyUTest;P<0.001) but no significant difference indNbetween male-biased genes and unbiased genes (Difference of 0.0019, Mann-WhitneyUTest;P = 0.026), as shown in Fig. 3. The difference indSbetween male-biased and femalebiased genes were low, as shown in Fig. 4. Hence, the elevateddN/dSvalue in female-biased genes are due to the higher increase in nonsynonymous substitution rate (dN) and not due to the decrease of synonymous substitution rate (dS).
As positive selection signals may be masked by genes with longer coding sequences, we useddN/dS>0.5 as a threshold for positive selections, as used by others (Nydam & Harrison, 2011; Swanson,Wong, Wolfner, & Aquadro, 2004). Using this cut-offvalue, we identified several candidate genes that are suggestive of positive selection event. These genes are:PODXL(dN/dS= 1.374),MYH7B(dN/dS= 0.956),SLC26A10(dN/dS= 0.693),TNNT1(dN/dS= 0.662),RAPGEF5a(dN/dS= 0.580),MH7l(dN/dS= 0.574) andCLK4a(dN/dS= 0.508).
More than 76%, 83% and 85% of the male-biased, female-biased and unbiased genes, respectively, have higher codon adaptation index than reference codon adaptation index. There were no significant differences in codon adaptation index between groups. We then extracted the gene expression level for each gene in sex-biased groups and performed a correlation analysis between gene expression level and codon adaptation index. We found a positive correlation (R= 0.396,P<0.01), as shown in Fig. 5. This is consistent with studies done inDrosophila melanogasterandEctocarpus spp., where codon adaptation index correlates strongly with gene expression levels (Connallon &Clark, 2011; Lipinska et al., 2015). When compared codon adaptation index between male and female-biased genes, no significant differences between these two groups were found.
4. Discussions
In this study, we presented a transcriptomic reanalysis in the skeletal muscle of male and female in hybrid tilapia (O. niloticusxO.mossambicus) using the latest reference genome, which allows us to understand the difference in sex-biased gene expressions between sexes.We obtained 19.1 million reads on average for each sample. This sequencing depth is similar to other transcriptomic analyses in teleost (Li,Lin, & Xia, 2017; Smith, Bernatchez, & Beheregaray, 2013). Here, we analyzed our RNA-seq data with the latest assembly, O_niloticus_UMD_NMBU (Conte et al., 2017). In this assembly, PacBio long reads were used to close the gaps as well as resolve complex repeats in previous assembly. This allowed us to map more than 92% of all reads onto the reference genome. Based on our highR2value between RNA-seq expression level and qPCR expression value, we conclude that our RNAseq DEG analysis is accurate. We detected 136 sex-biased genes in the skeletal muscle tissues with high confidence. From our results, we found that genes and pathways promoting growth are upregulated in males, while genes related to negative regulation of growth are upregulated in females. One of the pathways upregulated in males is theMAPKsignaling pathway.RASGRF1,an upstream gene in theMAPKsignaling pathway (Font de Mora et al., 2003), is highly expressed in male skeletal muscle tissue. InRASGRF1knock-out mice, somatic growth has been reported to be delayed.RASGRF1-deficient mice also showed hypoinsulinemia and low glucose tolerance due to a reduction of Beta-cells in the pancreatic islet, which produces insulin (Font de Mora et al., 2003). Insulin growth factor 1 (IGF1), which is a short peptide hormone associated with growth have been reported to be low in level inRASGRF1knock-out mice, hence causing slow growth in mice (Font de Mora et al., 2003). In tilapia, we detected high malebiasedRASGRF1expression, which may play a role in superior growth in male tilapia.
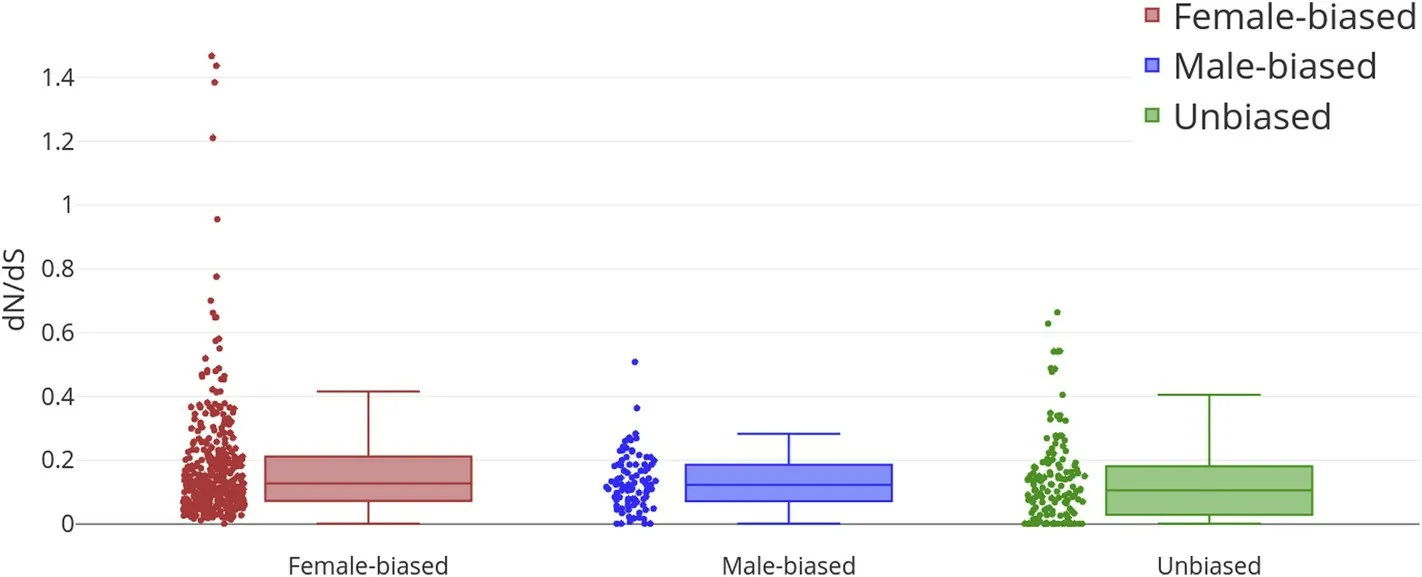
Fig. 2. Nonsynonymous to synonymous mutation rate ratio (dN/dS) of female-biased (N = 366), male-biased (N = 92) and randomly selected unbiased genes(N = 145). *** means P <0.01.
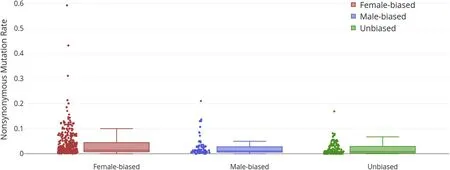
Fig. 3. Nonsynonymous mutation rates (dN) of female-biased, male-biased genes and unbiased genes. The median dN of female and male-biased genes were 17.4%and 13.5% higher than unbiased genes, respectively.
In another study of sexual size dimorphism inC. semilaevis,MAPKpathway was also found to be upregulated in females, which has faster growth rate compared to males (Wang et al., 2018). One of the pathways upregulated in females, is theFoxOsignaling pathway, which consists of a transcription factor family responsible for suppressing the expression of genes related to cell growth, proliferation and differentiation. One of its members, which we found to have a female-biased expression in tilapia skeletal muscle, isFoxO4.This gene has been shown to be an inhibitor of myogenic differentiation in muscle tissues,hence it is a negative regulator of somatic growth (Shi et al., 2012).Another gene from theFoxOsignaling pathway that is female-biased in expression is the insulin receptor substrate 2 b (IRS2b).IRS2b-/-mice have been shown to be 30% heavier in bodyweight at 32 weeks old,suggesting the role ofIRS2bin regulating growth and energy homeostasis (Lin et al., 2004).
Our study provided novel insights into sex-biased genes in skeletal muscle tissue, which is a somatic tissue. We showed that many genes(e.g.RASGRF1, FoxO4andIRS2b) are sex-biased in expression and may contribute to sexual size dimorphism, as these genes are key regulators of cell growth and proliferations. However, we would like to highlight that these genes are not located on chromosomes known to be associated with sex determining locus. Our results suggest that there may be other genetic factors in regulating sexual size dimorphism, besides sex determining genes. The mechanisms of achieving sex-biased gene expression in the presence of different sex chromosome remain largely unknown in tilapia. In future works, it may be essential to design an experiment to investigate how sex hormones (e.g. Testosterone) or sexdetermining genes (e.g.AMHandDMRT1) regulate somatic gene expression in fish. It could be also interesting to link sex determination,culture condition, DNA methylation (Wan et al., 2016) and sexual dimorphism.
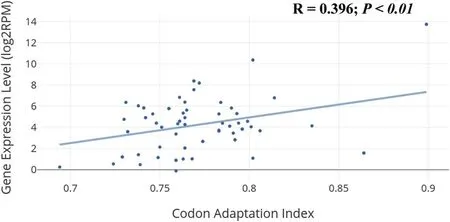
Fig. 5. Correlation between gene expression level (log2RPM) and codon adaptation index, which is positive and statistically significant (R = 0.396,P <0.01).
We also identified three sex-biased candidate genes that are located within a QTL for higher growth rate in hybrid tilapia (O. nilotiocusxO.mossambicus) (Lin et al., 2016). One of the genes isCADM3, which is involved in cell to cell adhesion activity. The second gene is a novel gene, LOC100706127, with no known function. It has a highly conserved immunoglobulin domain, suggesting that it may be involved in immune function. The third sex-biased gene located in this QTL is a non-coding gene, termed LOC109195677. It is located within the intronic region of the gene LOC100706127. The function of this ncRNA is unknown. In future studies, these three genes may be candidate genes for identifying DNA markers for performing association studies related to growth in tilapia and for markers-assisted selection for growth if the DNA markers are associated with growth. We reported significant associations of three SNPs in the promoter regions ofRASGRF1with bodyweight at 220 dph. This result suggests thatRASGRF1is a candidate gene related to growth rate in tilapia for future detailed functional investigations. These three SNPs may also be used for marker assisted selection for fast growth in tilapia breeding. However, it should be noted that these three markers may only be effective in our hybrid tilapia population as SNPs may be family or population specific. Further field trials using various other populations or strains are needed before these markers can be adopted for marker-assisted selection in other populations.
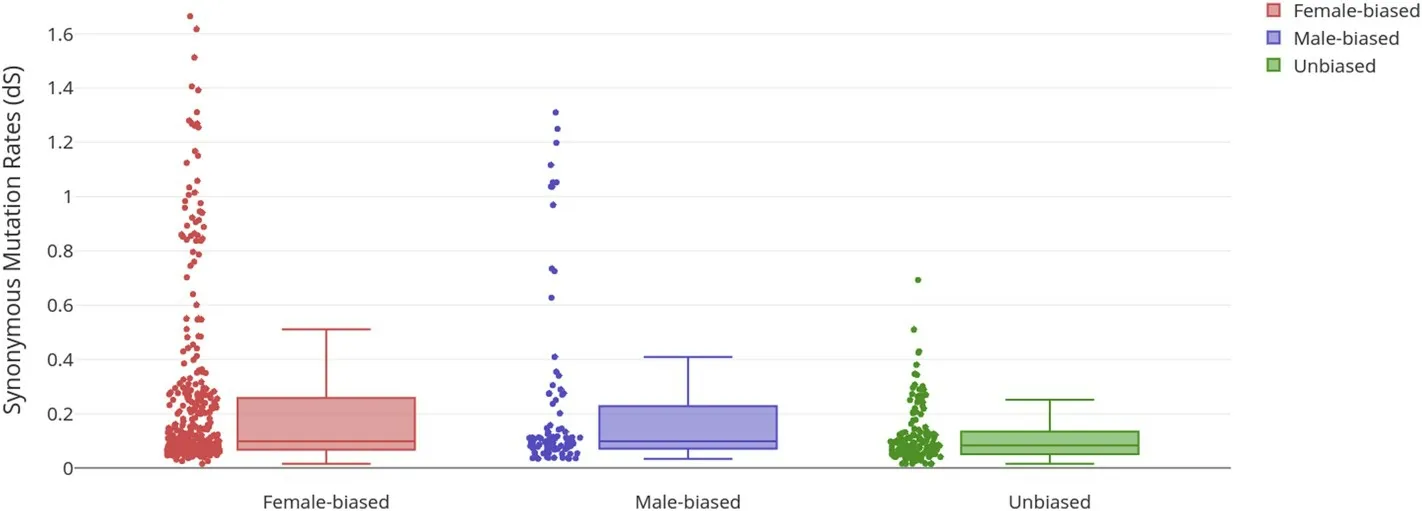
Fig. 4. Synonymous mutation rates (dS) of female-biased, male-biased genes and unbiased genes. dS of both female and male-biased genes showed significant increases (P <0.05) instead of decrease, hence suggesting that the higher dN/dS values are not due to the decrease in dS but are due to increase in dN.
Besides protein coding genes, we also identified six non-coding RNA genes in our list of sex-biased genes. These genes may play regulatory roles in fine tuning the expression levels of other genes via chromosomal inactivation, such as in the case ofXistwhich inactivates one of the X-chromosomes in female placental mammals (Reinius et al., 2010).However, the functions of these six non-coding RNAs are unknown. It is also interesting to note that although our libraries were constructed with oligo-dT enrichment methods, we were able to enrich some of the non-coding RNAs into our cDNA libraries. Studies have shown that a portion of known non-coding RNAs were known to have poly-A tails,hence it is possible for some non-coding RNAs to be enriched in oligodT enriched library(Cech & Steitz, 2014; Collins, Schönfeld, & Chen,2011). However, it should be pointed out that if one wishes to perform transcriptomic analysis targeted towards non-coding RNA, a ribosomal depletion method should be used instead (Arrigoni et al., 2016).
In this study, we also investigated the evolution rate of sex-biased genes in tilapia. Taking advantage of the publicly available reference genome of evolutionarily close species such as cichlids, we were able to calculate and estimate the evolution rate of each gene (Brawand et al.,2014). Based on our calculations,dN/dSof female-biased genes are 17.4% higher than unbiased genes. For male-biased genes,dN/dSis 13.5% higher than unbiased genes. These results suggest that some sexbiased genes in tilapia may be under positive selections. This result is consistent with other studies that investigate the evolutionary rate of sex-biased genes (Leder et al., 2010; Swanson et al., 2004; Yang, Zhang,& He, 2016). We also showed that this difference indN/dSratio is not due to decrease in synonymous mutation rate (dS) but is due to the increase in nonsynonymous mutation rate (dN). Although in our results,we showed that the median value ofdN/dSis higher for sex-biased genes, it is essential to note that only a subset of sex-biased genes is under positive selections, while the majority of sex-biased genes are under relaxed selection. We also showed that in teleosts, both male and female sex-biased genes are under selection pressure, which is consistent with results from zebrafish and stickleback studies (Leder et al.,2010; Yang et al., 2016). Studies have shown that genes with sex-biased expressions, such as those inDrosophila melanogaster, are rapidly evolving in the protein level (Connallon & Clark, 2011). Research done in avian species, fruit flies and zebrafish have also suggested that positive selection is the main driving force behind the rapid evolutions of sexbiased genes (Connallon & Clark, 2011; Ellegren & Parsch, 2007; Grath& Parsch, 2016; Leder et al., 2010). These positive selection events may be due to sexual selections or antagonistic coevolution between the sexes. Tilapia males are territorial, especially when defending a breeding nest and during breeding seasons (Chervinski, 1965). Hence,such reproductive cycle arrangements may lead to sexual selection for larger and faster growing males. We investigated the CAI of sex-biased genes and showed that there was a positive correlation between CAI and gene expression level (log2RPM), suggesting that sex-biased gene expression have been associated with optimal codon usage to promote efficient protein translation when mRNA is highly transcribed (Duret &Mouchiroud, 2000; Ellegren & Parsch, 2007; Lipinska et al., 2015).
In conclusion, we identified 136 sex-biased genes in skeletal muscles in hybrid tilapia. In male-biased genes, genes related to cell proliferation and growth are upregulated while in female-biased genes, genes related to growth inhibition and negative regulation were upregulated.We also showed three SNPs associated with bodyweight differences located atRASGRF1promoter region. These three SNPs may be used in marker-assisted selections for fast growth while theRASGRF1can be a candidate gene for future detailed research on its function on growth traits. Some of the sex-biased genes are novel non-coding RNAs and are interesting candidate genes for future functional analysis. We also showed that some sex-biased genes were under selection pressures and evolved in a faster rate compared to unbiased genes. The outcome of this study may be useful for candidate gene selection for future detailed functional analysis related to sexual size dimorphism using genome editing methods such as CRISPR/cas9 (Li et al., 2014). Candidate genes and SNP markers discovered in this study may be used for identifying DNA alleles associated with growth for marker assisted selection to accelerate genetic improvement of growth in hybrid tilapia.
Data availability
RNA-seq reads used in this study have been deposited into NCBI BioProject with the accession codes PRJNA309880.
Ethics statement
All handling of tilapia was conducted in accordance with the guidelines on the care and use of animals for scientific purposes set up by the Institutional Animal Care and Use Committee (IACUC) of the Temasek Life Sciences Laboratory, Singapore. The IACUC has approved this study within the project “Breeding of Tilapia” (approval number TLL (F)-12-004).
Author contributions
GHY initiated and coordinated the research project for ZYW. ZYW conceived and conducted the analysis. GL bred and raised the tilapia population used in this study. ZYW and GHY drafted and finalized the manuscript. ZYW and GHY assisted with experiments, data analysis and manuscript preparation.
Conflict of interest statement
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Acknowledgements
This research was supported by the National Research Foundation,Prime Minister's Office, Singapore, under its Competitive Research Program (CRP Award No. NRF-CRP7-2010-01) and the internal fund of Temasek Life Sciences Laboratory, Singapore. We thank Mr Ye Baoqing for manuscript proof-reading and Professors VCL Lin for supervising the PhD study of Zi Yi Wan.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.aaf.2019.05.003.
 Aquaculture and Fisheries2019年6期
Aquaculture and Fisheries2019年6期
- Aquaculture and Fisheries的其它文章
- A corpus-based approach to understanding market access in fisheries and aquaculture international business research: A systematic literature review
- Somatostatin 4 regulates growth and modulates gametogenesis in zebrafish
- Transcriptome analysis of gill from Lateolabrax maculatus and aqp3 gene expression
- A trap with light-emitting diode (LED) lights: Evaluating the effect of location and orientation of lights on the catch rate of snow crab(Chionoecetes opilio)
- Inter-individual variation in trophic history of Dosidicus gigas, as indicated by stable isotopes in eye lenses