
利用旁侧引物提高重叠延伸PCR 定点突变效率
2019-12-04 09:30:22王柳月李慧美马梦琪梁明星贺如阳陈华波
生物技术通报 2019年12期
王柳月 李慧美 马梦琪 梁明星 贺如阳 陈华波
(湖北文理学院医学院,襄阳 441053)
定点突变是研究基因功能与蛋白质结构的常用方法,目前主要使用PCR 介导的突变引入技术。PCR 即聚合酶链式反应,是一种在体外迅速扩增特定目标DNA 片段的分子生物学技术[1-2]。在此基础上发展起来的重叠延伸PCR(Overlap extension PCR,OE-PCR)是基因定点突变的主要方法[3-5]。整个OE-PCR 需要4 条引物,两轮共3 次PCR 反应,其繁琐的流程一直为科研工作者所诟病[6-8]。近年来陆续发展出新的定点突变解决方案,主要有滚环扩增法与大引物PCR 法[9],据称能简化流程,提高突变效率。也有研究者认为这两种方法各有弊端,如滚环扩增法容易引入新的非预期突变[10],而大引物PCR 法始终无法彻底避免对原始基因的扩增[11]。因此,大多数科研工作者仍倾向于使用OE-PCR 法制作定点突变,并不断提出优化方案以期提升工作效率。如有人提出平行模板法可以避免原始基因的扩增,于是可在同一个小管内完成对基因突变体的扩增[12]。
结合实际工作经验,我们发现OE-PCR 定点突变时效率不稳定,表现为转化之后的克隆形成数较少,克隆阳性率偏低。另一个较为普遍的现象是当把基因从一个载体中酶切出来直接连入另一个载体时,转化形成的克隆数较多,检测阳性率较高(几乎100%)。而克隆一个新基因或制作一个定点突变时,转化形成的克隆数较少,检测阳性率也往往不高。适当增加酶浓度,或延长酶切时间有助于增加克隆形成数和阳性率,但始终无法达到前者的效果。两者的主要区别在于前者酶切PCR 产物时切点往往位于DNA 末端,而后者酶切质粒时切点位于DNA 中间。大多数限制性内切酶都不能作用于DNA 顶端序列,因此在设计引物时会在酶切位点外侧增加1-3 个“保护碱基”,尽管如此其酶切效率往往只能达到最大值的20%-50%.据此推测靠近PCR 产物末端的低效限制性酶切是制约基因克隆和定点突变效率的主要原因。据此,我们提出旁侧引物法以期改善定点突变效率。即将常规OE-PCR 上、下游引物匹配位点由基因两端外延50-100 bp,适当增加第二轮PCR 产物的长度,使目标基因两侧的酶切位点远离DNA 末端,从而提高随后的酶切效率。
周期蛋白依赖性激酶4(Cyclin-dependent kinase 4,CDK4)是细胞周期的重要调节基因[13-14],它与cyclin D1以复合物的形式共同调节细胞周期G1/S 转换[15-16]。CDK4_D158N 是其重要的激酶活性缺失突变体,常用于CDK4功能研究[17]。以制作CDK4_D158N 为例,采用常规引物与旁侧引物法并行OEPCR 操作,发现旁侧引物法极大地提高了基因定点突变的克隆形成数与突变成功率。
1 材料与方法
1.1 材料
1.1.1 菌种与质粒 感受态大肠杆菌Trans5α Chemically Competent Cell(CD201-01)购自北京全式金生物技术有限公司;质粒pEGFP_C2(Catalog#6083-1,Clontech),pcDNA3.1(Catalog no. V800-20,Invitrogen)及CDK4基因相关质粒由北京大学生命科学学院张传茂教授馈赠。
1.1.2 试 剂 2×PCR Reagent(KT207) 购 自 天根生化科技(北京)有限公司;PyrobestTMDNA Polymerase(R005A),DNA Ligation Kit Ver.2.1(6002)购自Takara 宝生物工程(大连)有限公司;内切酶EcoR I(R0101V)、SalI(R0138V)购自NEB 纽英伦生物技术北京有限公司;DNA 快速回收/纯化试剂盒(NEP013-2)购自北京鼎国昌盛生物技术有限责任公司;质粒小量抽提试剂盒(D0003)购自碧云天生物技术。
1.2 方法
1.2.1 引物设计与合成 旁侧引物序列参考Invitrogen 中国测序通用引物序列(http://www.thermofisher.com/content/dam/LifeTech/Documents/PDFs/china/common-vector-primer-list.pdf),其他引物采用Primer Premier 5. 0 软件设计,相关引物序列见表1。引物由北京奥科鼎盛生物科技有限公司合成,经PAGE 纯化。

表1 引物信息表
1.2.2 PCR 扩增目标基因及阳性菌落检测 扩增目标基因采用高保真PyrobestTMDNA Polymerase,第一轮PCR 方案:2 μL 10×buffer,1.2 μL dNTP,0.2 μL primer 1,0.2 μL primer 2,0.2 μL 酶,0.1 μL(10 ng)质粒,补水至20 μL;第二轮PCR 方案:2 μL 10×buffer,2 μL dNTP,0.2 μL primer 1,0.2 μL primer 2,0.2 μL 酶,0.5 μL 上游片段回收物,0.5 μL下游片段回收物,补水至20 μL;运行程序皆为:94℃ 5 min;94℃ 30 s,54℃ 30 s,72℃ 1 min,30 循环;72℃ 10 min。PCR 产物经1%琼脂糖凝胶电泳检测后回收至20 μL ddH2O 中。用PCR 法检测阳性菌落时选用快速2×PCR Reagent,方案:5 μL 2×Taq,0.1 μL primer 1,0.1 μL primer 2,0.8 μL 细菌培养物,补水至10 μL;扩增条件:94℃ 5 min;94℃ 30 s,54℃ 30 s,72℃ 45 s,30 循环;72℃ 5 min。
1.2.3 基因克隆与序列分析 取5 μL 含点突变的目的基因回收物,加2 μL 10×buffer,0.5 μLEcoRI,0.5 μL,SalI,补水至20 μL,37℃孵育一定时间;取pEGFP_C2 质粒1 μg,采取上述同样方式酶切1 h。酶切产物经电泳检测后回收至20 μL ddH2O 中。用DNA Ligation Kit Ver.2.1 连接:载体0.5 μL,DNA片段4.5 μL,solution I 5 μL 混合,16℃孵育30 min。连接产物直接转化感受态大肠杆菌,用卡那霉素固体培养基筛选,结合PCR 检测阳性菌落。对相应PCR 产物回收或阳性菌落小量提取质粒后进行序列测定,序列测定采用ABI3730 系列测序仪,由北京奥科鼎盛生物科技有限公司完成。
2 结果
2.1 平行模板法无法避免原始基因的扩增
平行模板法即选用两个含同一目标基因的不同质粒作为模板,在这两个质粒上分别选一段序列作为引物[18],支持者认为“当两种模板和两条外侧引物同时存在于一个反应管时,可以完全避免对野生型目的基因全长的扩增[12],只有在突变引物存在的情况下才能扩增出产物,因此目标产物一定含有预期定点突变。为检测上述理论,选用pEGFP_C2-CDK4与pcDNA3.1-CDK4两种质粒作为平行模板,选用pcDNA3.1 多克隆位点(Multiple cloning site,MCS)上游86 bp 处的T7 promoter(pEGFP_C2 中无对应序列)作为上游引物,选用pEGFP_C2 MCS 下游61 bp 处的pEGFP_C3′(pcDNA3.1 中无对应序列)作为下游引物。将两种质粒单独作为模板时,都无法扩增出目标产物;将两种质粒混合物作为模板时,很好地扩增出目标产物(图1-A)。经序列测定,该PCR 产物中间为CDK4基因,两端多余序列分别源自pcDNA3.1 与pEGFP_C2(图1-B)。该DNA 片段长1 057 bp,恰好为86 bp,909 bp(CDK4基因除终止密码TGA 之外的长度),61 bp 之和(图1-C)。可见平行模板法无法避免对原始野生型基因的扩增。
2.2 两种方案下目标基因的扩增
为验证旁侧引物法的优点,以在pEGFP_C2-CDK4基础上制作其活性缺失突变体CDK4_D158N为例,采用常规末端引物与旁侧引物进行并行OEPCR 操作。常规末端引物分别为匹配CDK4基因两端序列的CDK4_S(含EcoR I 位点与3 个保护碱基)与CDK4_A(含SalI 位点与2 个保护碱基);旁侧引物pEGFP_C5′位于EcoRI 位点上游75 bp,旁侧引物pEGFP_C3′位于SalI 位点下游61 bp(图2-A)。
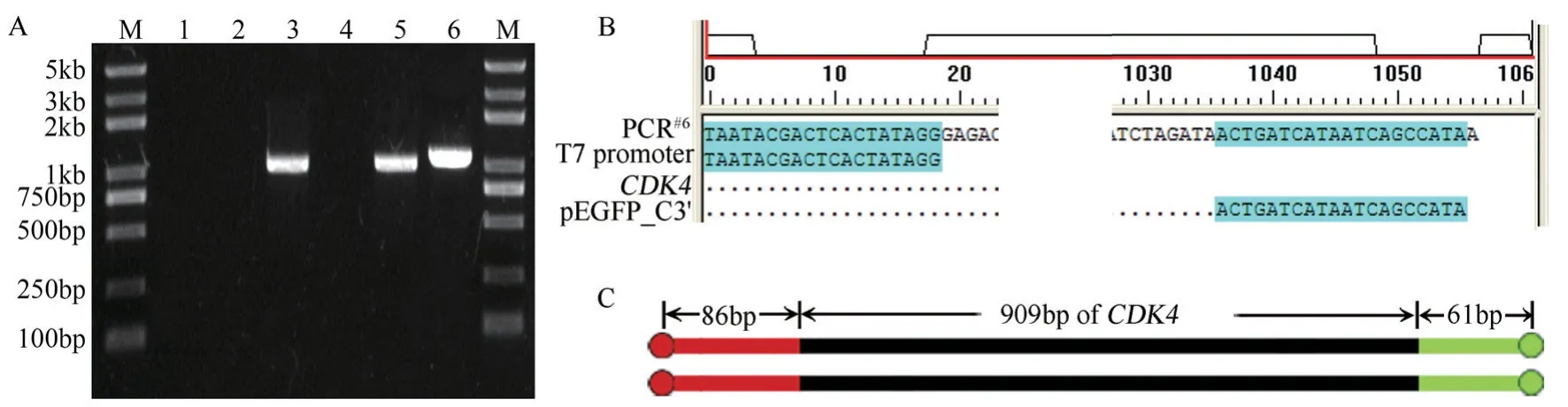
图1 平行模板法仍能扩增原始基因
第一轮PCR 产物经电泳检测,得到各自所需的4 个片段(图2-B)。将a+b 组合,c+d 组合进行第二轮PCR,两者皆得到相应的目标产物(图2-C)。且电泳结果显示采用旁侧引物时各PCR 产物都略大于常规末端引物下的产物,与预期相符。
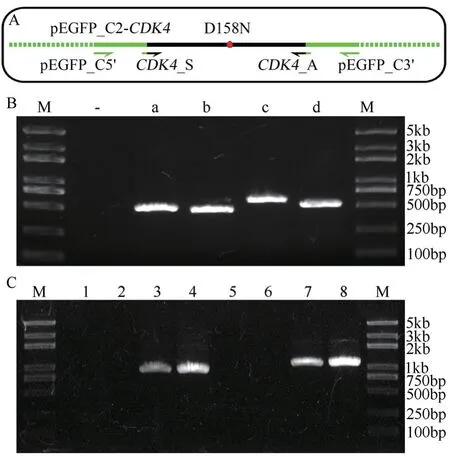
图2 OE-PCR 扩增含点突变的目标基因
2.3 突变体的克隆与鉴定
为检测不用酶切时间对常规末端引物法的影响,对上述3 号产物(图2-C)的双酶切设置1 h 与4 h两个时间组,以对比其效率差异。电泳结果显示不同酶切作用时间下产物大小基本一致,看不出三者的明显区别(图3-A)。由于此时识别位点位于DNA末端,双酶切只会使其缩短11 bp,故难以通过电泳结果判断酶切作用是否充分。
为检测旁侧引物法下切除两端多余序列后对终产物大小的影响,对7 号产物(图2-C)用EcoRI与SalI 以不同的组合方式进行酶切。考虑到此时识别位点位于DNA 中间,酶切效率高,故仅设置一个短时间组。电泳检测发现4 种酶切组合下的产物大小不同,其中双酶切产物最小,且条带单一(图3-B),说明该产物已被充分酶切。

图3 第二轮PCR 及酶切结果
将上述双酶切产物连入载体后经转化、培养以筛选目标突变体。常规末端引物下酶切1 h 组只长出3 个菌落,经PCR 检测,其中仅一个阳性克隆(图4-A),阳性率仅33%。常规末端引物下酶切4 h 组长出数十个菌落,任意挑取10 个菌落进行PCR 检测,得到7 个阳性克隆(图4-B),阳性率达70%。旁侧引物双酶切1 h 组长出数百个菌落,任意挑取10 个菌落进行PCR 检测,发现全部都是阳性克隆(图4-C),阳性率100%;对这10 个阳性克隆提取质粒进行序列测定,结果显示全部都是pEGFP_C2-CDK4_D158N,即突变成功率为100%。
3 讨论

图4 不同方案下的平板筛选及菌落检测结果
由于OE-PCR 定点突变程序繁琐,很多科研工作者都试图提出优化方案,平行模板法即其中之一。然而,我们发现平行模板法并不能避免对原始野生型基因的扩增。结合具体实例,并不难分析其原理。由T7 promoter 与pEGFP_C3′引物各自完全延伸的单链DNA 虽然在CDK4区域互补,但是没有配对的3′-OH 末端,无法继续延伸。而PCR 反应中往往会有部分未完全延伸的产物,如果两者恰好在各自的CDK4序列内部中止延伸,那么这两条单链DNA不只互补,且各有一个配对后的3′-OH 末端。因此可以互为模板继续延伸,形成一条“杂交”DNA 双链。其上游序列源自pcDNA3.1,中部为CDK4序列,下游序列源自pEGFP_C2。该“杂交”DNA 双链可以作为后续循环的模板使目标基因CDK4得以扩增(图5)。因此,试图以平行模板法在一个小管内完成OE-PCR 所有扩增反应的构思是不成立的。
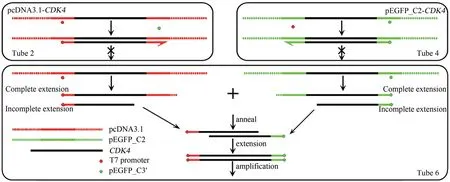
图5 平行模板法扩增目标基因原理示意图
Ⅱ型限制性内切酶识别DNA 的特定序列并切开双链DNA,若酶切位点靠近DNA 末端,则酶切效率受到不同程度的影响[19-20]。在设计引物时,往往在酶切位点外侧添加1-3 个“保护碱基”以保证PCR 产物的酶切效率。尽管如此,其酶切效率有时也只能达到最大值的20%-50%(图6-A)。低效酶切也成为基因克隆尤其是PCR 定点突变的重要制约因素。有两种方法可提高此时的酶切效率,一是增加引物中“保护碱基”数目,但相应地会增加实验费用,且非配对碱基数的增加还会降低PCR 成功率;二是增加酶切反应的酶量或(和)延长酶切作用时间,却会增加内切酶出现星号活性的风险。选用旁侧引物可以很好地解决上述问题,且可以避免诸多不利因素。旁侧引物位于酶切位点外侧50 bp-100 bp,此时对PCR 产物酶切近似于甚至优于从质粒中切取基因片段(质粒的超螺旋结构会影响酶切效率),极大地提高酶切反应的效率(图6-B),由此缩短反应时间,增加克隆形成数及阳性率。

图6 旁侧引物法提高PCR 产物酶切效率原理示意图
除此之外,旁侧引物法还有以下两个优点,一是双酶切产物与非酶切或单酶切产物大小有一定差异,可以通过电泳结果直接判断酶切是否充分(图3-B)。另外,当突变位点靠近基因两端时,采用常规末端引物会使第一轮PCR 产物之一偏小,不便于检测及回收。此时采用旁侧引物可以适当增加DNA片段长度,便于后续操作。旁侧引物源于质粒载体上的一段序列,只要插入该载体的基因,都可以之制作定点突变,不会过多增加实验成本。
平行模板法虽无法避免原始基因的扩增,但是其扩增效率明显低于同一个质粒上、下游引物对目标基因的扩增。因此,我们认为仍可利用两个平行质粒分别作为第一轮PCR 上下游片段的扩增模板,以减少定点突变过程中原始质粒对第二轮PCR的“污染”。如果在第一轮PCR 时选用两种不同的平行质粒,除非两者同时污染各自的PCR 产物,否则第二轮PCR 不会扩增出原始基因,这样就极大地减少了原始质粒对后续操作的污染。
4 结论
本研究论证了平行模板法简化OE-PCR 定点突变流程的思路不能成立。我们认为制约OE-PCR 定点突变效率的核心因素是靠近DNA 末端识别位点的低效限制性酶切。采用旁侧引物法可提高PCR 产物的酶切效率,且便于通过电泳监测酶切效果,既节省工作时间,又能大幅提高OE-PCR 定点突变的成功率。
猜你喜欢
环球时报(2022-09-20)2022-09-20 15:18:57
中学生数理化(高中版.高考数学)(2022年4期)2022-05-25 13:07:02
今日农业(2021年21期)2021-11-26 05:07:00
新世纪智能(教师)(2021年2期)2021-11-05 08:43:20
教育周报·教育论坛(2021年21期)2021-04-14 00:09:18
今日农业(2020年24期)2020-12-15 16:16:00
食品科学(2018年10期)2018-05-23 01:27:28
兽医导刊(2016年12期)2016-05-17 03:51:50
西南医科大学学报(2015年1期)2015-08-22 13:01:46
中国当代医药(2015年9期)2015-03-01 02:01:59
