
超高效液相色谱-串联质谱法检测肉丸中诺氟沙星药物残留
2019-12-02 08:02程海霞
中国科技纵横 2019年16期

摘 要:采用甲酸化的乙腈溶液提取肉丸中诺氟沙星药物,并采用固相萃取净化提取液,建立了肉丸中诺氟沙星的超高效液相色谱-串联质谱检测方法。样品经2%甲酸乙腈提取,提取液盐析,取乙腈层过固相萃取柱净化,在电喷雾离子源正离子模式下,多反应离子监测进行测定,内标法定量。诺氟沙星药物在2.0~50 ng/mL范围内线性关系良好,相关系数为0.9997。在3个不同添加水平下的平均回收率为87.0~91.0%,相对标准偏差为6.2~7.1%(n=6),检出限为0.5 μg/kg。该方法操作简便、快速,净化效果好,灵敏度高,适用于肉丸中诺氟沙星药物残留的快速分析检测。
关键词:固相萃取;超高效液相色谱-串联质谱法;诺氟沙星;肉丸
中图分类号:TS251 文献标识码:A 文章编号:1671-2064(2019)16-0000-00
诺氟沙星是第三代喹诺酮类抗菌药物中的一种,该药物为广谱抗菌抗菌药,其见效快,且成本低,因此被广泛的使用到动物的养殖中[1],为畜禽和水产专用抗菌药物,但部分养殖户过度使用该类药物,或未注意休药期,导致畜禽和水产产品中该类药物残留较多,同时当前对于肉丸类食品中该种残留没有限量指标,因此建立肉丸中诺氟沙星的检测方法具有重要的实际意义。
目前,食品中诺氟沙星的检测方法主要有液相色谱法[2]、液相色谱-质谱联用法[3-6]。本文以市售的肉丸为研究对象,对其诺氟沙星药物检测的样品前处理条件进行了选择和优化,超高效液相色谱-串联质谱检测器检测,对液相分离条件和质谱检测条件分别进行了优化,建立了超高效液相色谱-串联质谱检测肉丸中诺氟沙星的检测方法,并对市售的10批次实际样品进行了检测。
1 实验部分
1.1 仪器与试剂
WATERS TQD质谱仪(美国WATERS公司);XEVO超高效液相色谱仪(美国WATERS公司);BEH Shield C18色谱柱(50 mm×2.1 mm,1.7μm,美国WATERS公司);高速冷冻离心机(美国热电公司); Milli-Q超纯水仪(美国Millipore公司);氮吹仪(上海屹尧仪器科技发展有限公司);涡旋混合仪(德国IKA公司); Oasis HLB固相萃取柱(美国WATERS公司)。
甲醇、乙腈均为色谱纯(美国Fisher公司);甲酸,色谱纯(上海安谱科学仪器有限公司);氯化钠、氨水均为分析纯,购自国药集团化学试剂有限公司;水为Milli-Q超纯水仪制备的超纯水。
标准品诺氟沙星购置于中国食品药品检定研究院,纯度为99.5%。
1.2标准溶液的配制
精密称取诺氟沙星标准品适量,加入0.2 mL甲酸,用甲醇溶解并定容至刻度,配制成质量浓度约是1.0 mg/mL标准储备液,置于棕色瓶中,于冰箱-18℃中保存。用时根据需要,用0.1%甲酸-乙腈(95:5)溶液逐级稀释为标准工作液,临用现配。
1.3样品处理
试样经均质器均质后,称取约5 g(精确至0.01 g)于50 mL离心管中,加入15 mL2%甲酸乙腈溶液,涡旋混合30 s;8000 r/min离心10 min,将上清液转移至另一离心管中,残留物再用10 mL1%甲酸乙腈溶液提取两次,合并三次提取液;加入10 mL乙腈饱和的正己烷,涡旋30 s,取乙腈层于氮气流下吹至近干,加入5 mL水,涡旋混合,10000 r/min的速度离心5 min待净化。
将样品溶液转移至固相萃取柱中(固相萃取柱依次用6 mL甲醇、6 mL水活化),以6 mL水洗涤小柱,抽干后,用乙腈-1 mol/L氨水(95:5)溶液6 mL洗脱,收集洗脱液于氮气流下吹至近干,用初始比例流动相1.00 mL复溶,涡旋混合,过0.22 μm有机滤膜供UPLC-MS/MS分析测定。
1.4基质标准溶液配制
取空白基质麻辣烫样品,按1.3方法处理至浓缩至近干,用标准工作液1.0 mL复溶,涡旋混合30 s,过0.22 μm滤膜,即得。
1.5 色谱和质谱条件
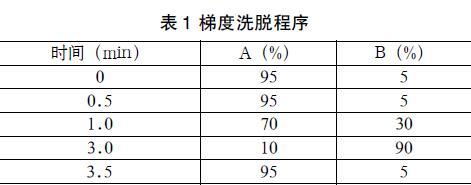
色谱柱:BEH Shield C18色谱柱(50 mm×2.1 mm,1.7μm);柱温:30 ℃;进样量:5 μL。流动相A为0.1%甲酸水溶液,流动相B为乙腈溶液;流速:0.35 mL/min。梯度洗脱程序见表1。
电喷雾离子源(ESI);正离子扫描;多反应离子监测(MRM);喷雾电压:2 kV;雾化气温度:400 ℃;雾化气流速:0.8 L/min;质谱采集参数见表2。
2 结果与讨论
2.1 色谱条件的优化
本研究中先后考察了乙腈-水和乙腈-0.1%甲酸水溶液作为流动相时,诺氟沙星的响应情况。结果表明,采用乙腈-0.1%甲酸水溶液作为流动相时,诺氟沙星的离子化效率有所提高,且峰形较好,因此乙腈-0.1%甲酸水溶液作為流动相;后又考察了不同浓度的甲酸水溶液对诺氟沙星的响应影响情况,发现变化不大,因此最终选择乙腈-0.1%甲酸水溶液作为流动相。
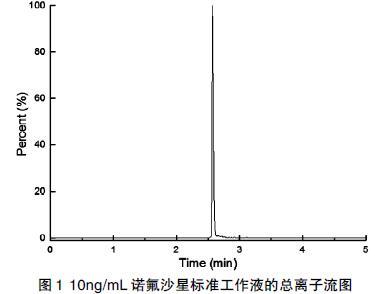
采用选定的流动相和色谱柱,对标准工作液进行检测,优化梯度洗脱程序,使诺氟沙星具有较好的峰形和灵敏度。图1为10ng/mL诺氟沙星标准工作液的总离子流色谱图。
2.2质谱条件优化
通过质谱端直接进样的方式将500 ng/mL的诺氟沙星标准工作液引入质谱检测器,分别进行正离子和负离子全扫描分析,发现诺氟沙星的最佳准分子离子峰在正离子扫描模式下获得。以其准分子离子为母离子,不断改变锥孔电压,选择准分子离子响应最高的电压作为锥孔电压;再通过改变碰撞电压,将准分子离子打碎,并检测子离子,选择稳定且响应最好的两个子离子作为特征子离子,其中丰度相对较强的为定量离子,另一个作为定性离子,再对两个特征子离子的碰撞能量进行分别优化,使定量离子和定性离子的响应均达最优。优化得到的质谱参数见表2。
2.3樣品前处理条件的优化
肉丸样品基质比较复杂,含有较高的蛋白、脂肪,同时还有不同含量的调味品等,因此对样品中的诺氟沙星提取后,要采用一定的净化手段去除大量的杂质,以期得到较为干净的待测液和较好的检测灵敏度。对于提取溶剂,我们选择了现有研究中常用的酸化乙腈溶液,并考察了添加不同体积分数的甲酸乙腈溶液的提取效果,实验表明2%甲酸乙腈溶液作为提取液即可达提取效率最优。肉丸样品中含有一定量的油脂,提取时也一并提取到提取液中,对后续的净化和检测均有影响,因此选择乙腈饱和的正己烷对提取液进行除油操作。
2.4 线性方程、检出限
按照1.4项的方法配制5个水平的基质标准溶液,并采用优化的色谱和质谱条件进行检测。以诺氟沙星定量离子的峰面积(y)作为纵坐标、质量浓度(x,μg/L)为横坐标绘制标准曲线,线性范围为2.0~50 μg/L,相关系数r为0.9997,线性关系良好,检出限为0.5 μg/kg。
2.5回收率和精密度
在空白肉丸样品中添加诺氟沙星标准溶液,制备含量分别为1.0、2.0和5.0 μg/kg的加标样品,按照前处理方法及优化的检测条件进行试验,每个添加水平平行测定6份,结果见表3,平均回收率为87.0~91.0%,相对标准偏差为6.2~7.1%。
2.6 实际样品检测
采用本文建立的方法,对10批次市售肉丸食品进行检测,9批次均未检出,1批次检出含有诺氟沙星,含量为7.6 μg/kg。
3结语
本文采用酸化乙腈溶液对肉丸食品中的诺氟沙星进行提取,并使用HLB固相萃取柱净化提取溶液,分别优化了诺氟沙星检测的液相色谱分离和质谱检测条件,建立了UPLC-MS/MS检测肉丸中诺氟沙星药物的分析方法,并将该方法用于市售肉丸食品的检测中。该方法操作简便、快速,其灵敏度较高,回收率和精密度均符合检测技术的要求,可用于肉丸食品中诺氟沙星的检测。
参考文献
[1] 兰海.浅谈喹诺酮类药物的发展与合理应用[J].疾病监测与控制杂志,2014,8(10): 649-650.
[2] 祝曙华,苏青云.四种固相萃取柱在猪肉四种氟喹诺酮类药物残留检测中应用比较[J].色谱安全质量监测学报,2014,5(2):415-419.
[3] 刘辉,谭素娴.固相萃取-超高效液相色谱-串联质谱法测定水产品中多种兽药的残留量[J].理化检验(化学分册),2014,50(4):439-444.
[4] 郭霞,孙建华,孙振中,刘菁华,黄雪玲,刘云璐,陈琳.水产品中喹烯酮和喹赛多及其主要代谢物的HPLC-MS/MS检测方法研究[J].分析测试学报,2016,35(12):1535-1541.
[5] 张颖颖,李莹莹.超高效液相色谱-串联质谱测定猪肉中16种喹诺酮药物残留量[J].肉类研究,2016,30(5):36-41.
[6] 张科明,梁飞燕,邓鸣,刘向红,许杨彪,赵庄.QuEChERS结合液相色谱-串联质谱法快速测定猪肉中多类兽药残留[J].色谱,2016,34(9):860-867.
收稿日期:2019-07-05
作者简介:程海霞(1988—),女,汉族,湖北武汉人,硕士,实验工程师,助理实验师,研究方向:食品化妆品农兽药残留检测。
Determination of Norfloxacin in Meat balls using Ultra Performance Liquid Chromatography-Tandem Mass Spectrometric
CHEN Hai-xia
(Wuhan Customs Technology Center,Wuhan Hubei 430050)
Abstract:An ultra performance liquid chromatography-tandem mass spectrometric (UPLC-MS/MS) method was established to determination of the norfloxacin in meat balls. Samples were extracted with acidified acetonitrile and separated on SPE column, and analyzed by UPLC-MS/MS under the positive mode using multiple reaction monitoring(MRM). The method showed good linearities over the range of 2.0~50 ng/mL with good linear correlation coefficients (r=0.9997). The method detection limit of the norfloxacin was 0.5 μg/kg, and the recovery of norfloxacin in meat balls ranged from 87.0~91.0 % with the relative standard deviations (RSD) of 6.2~7.1% (n=6). The results indicated that this method is simple, fast and clean, and suitable for the simultaneous determination for the norfloxacin in the meat balls.
Key words:SPE; Ultra Performance Lquid Chromatography-tandem Mass spectrometric (UPLC-MS/MS); Norfloxacin; Meat balls
猜你喜欢
