
CRISPR/Cas9系统介导的miR-155基因敲除细胞的制备
2019-11-21 11:09:28李聪聪张永辉赵婉霞吴姣韩浩园李梦云牛晖宋素芳李婉涛
生物技术通报 2019年11期
李聪聪 张永辉 赵婉霞 吴姣 韩浩园 李梦云牛晖 宋素芳 李婉涛
(1. 河南牧业经济学院动物科技学院,郑州 450046;2. 河南省畜禽遗传资源保护工程技术研究中心,郑州 450046;3. 郑州市动物生殖分子调控重点实验室,郑州 450046)
CRISPR/Cas系统是由RNA介导的对DNA修饰的基因编辑工具,该技术可以对基因组特定位点进行缺失、插入、修复等靶向编辑,是目前基因组编辑领域最受欢迎的新技术。CRISPR/Cas系统是人们在古细菌和细菌中发现的一种获得性免疫机制。该系统有 3种类型:TypeⅠ、TypeⅡ和TypeⅢ[1],TypeⅠ和TypeⅢCRISPR/Cas系统需要多个Cas蛋白形成复合体切割DNA双链,其中仅存在于细菌的TypeⅡ系统只需要一个Cas9蛋白来切割DNA双链,而且体外实验已经证明Cas9基因是参与CRISPR免疫系统唯一的必须基因[2-3],由于其简易性和可操作性,TypeⅡ系统已被改造为最成功的人工核酸酶[4],最有力的基因编辑工具,成为基因编辑领域最炙手可热的技术。Cas9是一种内切酶,具有RuvC-like结构域和HNH核酸酶结构域两个内切酶活性中心。Jinek等[5]发现Cas9对双链DNA的切割作用,需要crRNA(CRISPR-derived RNA)和tracrRNA(trans-activating RNA)的介导。具体来说:crRNA和tracrRNA通过碱基互补配对原则结合形成一个嵌合RNA分子,被称为向导RNA(single guide RNA,sgRNA)。这个sgRNA与Cas9相互结合形成蛋白-RNA复合体,该复合体中的Cas9蛋白在crRNA引导序列靶位点剪切双链DNA,其中HNH核酸酶结构域切断与crRNA互补的一条链,RuvC-like结构域切断非互补链,形成DSB(DNA doublestrand breaks)[4],引起细胞的 NHEJ修复机制。该过程即是CRISPR/Cas9系统介导的基因组编辑分子基础。
miR-155促进机体免疫细胞分化,调控机体炎症反应和免疫应答,在机体抵抗感染中发挥重要作用。miR-155基因是重要的抗病候选基因。大量研究表明,miR-155在机体造血细胞分化、炎症反应以及免疫应答等生物过程中发挥重要作用[6]。miR-155是对免疫细胞的活化及炎症因子的表达具有调控作用的内源性非编码小分子RNA。Rodriguez等[7]通过miR-155敲除小鼠模型研究发现,miR-155是机体维持正常免疫活动所必须的。MiR-155通过在活化的B细胞和T细胞中上调表达来调节机体获得性免疫反应。在B细胞成熟过程中,miR-155靶向调控c-Maf、PU.1和AID等基因,促进记忆性B细胞的形成[8]。MiR-155通过靶向 SOCS1和 c-Maf,调控Th1/Th2分化平衡的机制[9]。MiR-155参与调控Th17和Th1细胞亚群的分化[10-11],而且在慢性移植排斥反应中,miR-155调控Th1/Th17相关的免疫反应[12]。MiR-155参与CD8+T细胞反应,miR-155缺失可抑制CD8+T细胞应答,miR-155超表达可增强CD8+T细胞应答[13]。MiR-155通过TLR信号通路途径可被细菌(LPS)和病毒(Poly I:C)的复合物诱导表达,也可被TNFα和干扰素(IFN-β,IFN-γ)诱导表达[14-17]。一些参与TLR诱导的信号级联放大的基因 FADD、IKKε、Ripk1、TAB2、SOCS1都是 miR-155 的靶基因[7,18-20]。这些研究表明 miR-155在机体先天性免疫中发挥重要作用,即miR-155调控机体炎症。大量研究表明miR-155的表达水平与炎症因子的表达水平相关联,是个亲炎症(Proinflammtory)分子。本课题组用LPS、Poly(I∶C)分别刺激PK15细胞,发现miR-155对TLR3/4发挥正调控作用[21]。另有研究用土拉弗朗西斯菌(Francisella tularensis)感染人单核细胞,检测到miR-155表达显著增加,并且上调表达的miR-155对促炎因子TNFα、IL1β、IL6的生成发挥正调控作用[22]。在类风湿性关节炎患者的外周血中,miR-155的表达水平与炎症因子TNF-α、IL1β的释放存在正相关[23]。前人及我们的研究发现miR-155不但调控多种免疫细胞成熟、分化,而且当机体受细菌或病毒侵染时,miR-155迅速上调以激活机体炎症反应,清除病原。Wang等[24]用RNA病毒感染小鼠巨噬细胞发现,miR-155上调表达,并靶向抑制SOCS1表达,进而增强I型干扰素信号通路,达到减弱病毒复制的目的。在研究人类艾滋病中,Poly(I∶C)刺激单核细胞来源的巨噬细胞时,高表达的miR-155能够抑制HIV-1的感染,也即miR-155在机体对抗病毒过程中发挥积极作用[25]。以上研究表明miR-155是重要的抗病候选基因。
存在机体组织微环境中的巨噬细胞是重要的免疫细胞,具有吞噬、抗原加工与递呈、免疫调节等生理功能,在机体先天性免疫和获得性免疫中发挥重要作用。猪体受到细菌(猪沙门氏菌、副猪嗜血杆菌)或病毒(猪蓝耳病)感染后,常常首当其冲受到影响的是巨噬细胞,如肺泡巨噬细胞。大量研究数据表明细菌(如副猪嗜血杆菌)感染削弱巨噬细胞的免疫力导致猪对细菌易感[26],病毒(猪蓝耳病)感染抑制巨噬细胞的功能形成严重免疫抑制导致猪体对其它病毒性疾病和细菌性疾病易感性明显增加[27]。本研究选定重要的抗病候选基因miR-155,采用改良的CRISPR/Cas9系统构建miR-155敲除的猪肺泡巨噬细胞系,为探讨miR-155在巨噬细胞发挥的调控功能研究提供细胞模型。
1 材料与方法
1.1 材料
pGL3-U6-sgRNA-PGK-puromycin(Addgene:#51133)和pEGFP-C1-cas9为华中农业大学谢胜松副教授课题组馈赠,3D4/21猪肺泡巨噬细胞由华中农业大学赵书红教授课题组馈赠。引物合成和测序均由生工生物工程(上海)股份有限公司完成。
1.2 方法
1.2.1 sgRNA设计及表达载体构建 利用sgRNAcas9软件在ssc-miR-155前体区域设计4条特异性的sgRNA引物,引物名称分别为sgRNA39F/R、sgRNA42F/R、sgRNA51F/R、sgRNA52F/R( 表 1),以 pGL3-U6-sgRNA-PGK-puromycin(Addgene:#51133,简写pGL3-U6-sgRNA)载体为骨架构建表达载体。提取包含4条sgRNA靶位点及ssc-miR-155前体序列在内的696 bp基因组序列,用Premier5.0软件设计最佳的PCR扩增引物对ssc-miR-155F/R(表1),引物合成由生工生物工程(上海)股份有限公司完成。sgRNA表达载体构建步骤为:取浓度为10 μmol/L的前向与反向引物各5 μL在PCR仪上用 95℃,10 min;65℃,1 h;pGL3-U6-gRNA质 粒用BsaI酶(NEB北京)进行酶切,酶切回收备用,连接退火后的sgRNA;将退火后的sgRNA稀释100倍,取1 μL与酶切的pGL3-U6-gRNA质粒,使用T4 DNA连接酶(宝生物工程(大连)有限公司)16℃连接1 h;取2 μL连接产物,转化到DH5α感受态细胞(宝生物工程(大连)有限公司)中,涂布到加了氨苄的LB固体培养基上,挑选用PCR扩增(此时用U6左引物和sgRNA右引物鉴定阳性克隆)鉴定成功的单克隆菌液,送生工生物工程(上海)股份有限公司用“U6seqF”引物测序鉴定,将靶向ssc-miR-155前体的不同位置sgRNA表达载体分别命名为:sgRNA51、sgRNA52、sgRNA42和 sgRNA39。测序正确的阳性菌扩大培养,用无内毒素质粒提取试剂盒(Endo-free Plasmid DNA Mini Kit Ⅱ,Omega Bio-Tek)抽提表达质粒,测定浓度后备用。
1.2.2 细胞培养,质粒转染及流式分选 3D4/21细胞培养,用含10%胎牛血清的1640(Gibco)培养基,在CO2浓度为5%,温度为37℃的培养箱中培养。转染前接种一个6孔板,每孔接种细胞量为1.2×105个,待细胞处于对数生长期且密度接近80%左右时,按照Lipofectamine®2000转染试剂说明书分组转染(对照组:pGL3-U6-sgRNA空载+ pEGFP-C1-cas9;实验组:pGL3-U6-sgRNA39/42/51/52+ pEGFPC1-cas9;第三组:pEGFP-C1-cas9),于转染48 h后收集所有细胞。若进行流式分选GFP阳性细胞,于转染24 h后收集所有细胞,转入流式细胞仪(BD FACSJazz,美国)进行分选,分选后的细胞添加含有1%双抗的完全培养基继续培养48 h收集所有细胞。利用基因组DNA小量抽提试剂盒(天根生化科技(北京)有限公司),抽提不同实验组的细胞基因组DNA。
1.2.3 PCR扩增与T7EI酶酶切鉴定、测序分析 以上述抽提的细胞DNA为模板,利用表1中的引物ssc-miR-155F/R,在Q5超保真 DNA 聚合酶(NEB,北京)作用下进行PCR扩增,PCR反应条件为:98℃预变性30 s,98℃变性10 s,64℃退火复性30 s,72℃延伸30 s,扩增30个循环。PCR产物用1.5%琼脂糖凝胶电泳检测。利用PCR纯化试剂盒(天根生化科技(北京)有限公司)纯化PCR扩增产物,接下来严格按照T7EI核酸酶(NEB,北京)的说明书进行操作:取200 ng纯化之后的PCR产物,加入2 μL NEBuffer 2,补充无核酸酶水至19μL,在PCR仪上进行梯度退火杂交,程序为:95℃变性5 min;95-85℃,每秒降低2℃(-2℃/s);85-25℃,每秒降低 0.1℃(-0.1℃/s);取 1 μLT7EI核酸酶,在PCR仪上37℃切割杂交的PCR产物15 min,然后在2%琼脂糖凝胶上电泳检测,基因组突变的泳道将会出现小分子量条带。进一步,取纯化后的PCR扩增产物,进行加A反应(天根生化科技(北京)有限公司),而后取加A后产物与pMD-19T载体(宝生物工程(大连)有限公司)进行TA克隆,DH5α感受态细胞(宝生物工程(大连)有限公司)转化,涂平板,挑选单克隆在LB培养基中扩大培养,菌液PCR鉴定为阳性克隆后送工生物工程(上海)股份有限公司进行测序验证。
1.2.4 细胞RNA提取及qRT-PCR 取1.2.2收集的 细 胞, 加 入 TRIzol(Invitrogen,Thermo Fisher Scientific,USA),按照RNA提取步骤提取每个样品的总RNA,测定浓度后备用。用DNAseⅠ(Invitrogen,Thermo Fisher Scientific,USA)对提取的总RNA去 除 基因组 DNA,反应体 系:1 μg总 RNA,1μL10×Reaction Buffer,1 μL(1U)DNase Ⅰ(RNasefree),补DEPC水至总体积10 μL,37℃反应30min。加 入 1 μL 50 mmol/L EDTA, 在 PCR 仪 上 65℃ 孵育10 min,终止反应。按照反转录试剂盒K1622(Invitrogen,Thermo Fisher Scientific,USA)的操作步骤进行反转录,cDNA合成步骤:1 μg去除基因组DNA后的总RNA,1 μL茎环引物(RT-ssc-miR-155-5p/ RT-ssc-miR-155-3p),补 Nuclease-Free Water 至12 μL 构成mix1,65℃孵育5 min,而后立即置于冰上 2 min ;4 μL 5×Reaction Buffer,1μL RiboLockTM RNase inhibitor,2 μL 10 mmol/L dNTP mix,1 μL RevertAidTM M-MuLV Reverse Transcriptase,将mix1和mix2混匀离心,设置反应程序:42℃,60 min;70℃,5 min;15℃,2 min。 反 应 结 束,-20℃ 保存。以反转录所得cDNA为模板,对ssc-miR-155-5p,ssc-miR-155-3p表达量进行检测分析,各基因检测引物见表1。qPCR 反应体系 :7.5 μL 2×SYBR®Green Real-time PCR Master Mix(Toyobo, 上 海 ),50 pmol引 物,75 ng模 板 cDNA, 补 水 至 15 μL。ABI7500 Fast 系统进行实时荧光定量PCR检测。扩增程序 :95℃,5 min(Holding stage);95℃,15 s;60℃,34 s;72℃,15 s(Cycling stage,40 cycles);95℃,30 s;58℃,34 s;95℃,15 s;60℃,15 s(Melt Curve Stage)。
1.2.5 Western blotting 取1.2.2所收集细胞,加RIPA裂解液(强)+1mmol/L PMSF(Phenylmethanesulfonyl fluoride,苯甲基磺酰氟)(碧云天,上海)裂解细胞。选用8%SDS-PAGE胶,上样量为30μg。本实验所用一抗有SHIP1(bs-3567R,博奥森Bioss),抗体稀释比 1∶500;beta-Actin(bsm-33036M,博奥森 Bioss),抗体稀释比1∶1000;荧光二抗StarBrightTMBlue 700 Goat Anti-Rabbit IgG(#12004162,伯乐),抗体稀释比为1∶10000;DyLight 800 Goat Anti-Mouse IgG(#STAR117D800GA,伯乐),抗体稀释比为1∶5000。最后在Bio-Rad ChemiDoc MP全能型成像系统中显色。采用Bio-Rad Image Lab software进行图像分析。
1.2.6 数据分析 qPCR数据由相对定量2-△△Ct法进行分析[28],U6作为内参基因对各基因表达水平进行校正。用非配对的t检验分析数据差异显著性,结果以平均值±标准差表示。
2 结果
2.1 合成 sgRNA引物并构建载体
根据在线CRISPR设计结果,合成评分较高的sgRNA引物序列(表1),pGL3-U6-sgRNA重组载体信息(图1),成功构建靶向ssc-miR155的pGL3-U6-sgRNA载体。
2.2 miR-155基因敲除的3D4/21细胞的制备
正常培养3D4/21细胞至密度为80%左右,用Lipofectamine®2000转染试剂进行分组转染,pGL3-U6-sgRNA39/42/51/52+pEGFP-C1-cas9为实验组;pGL3-U6-sgRNA空载+ pEGFP-C1-cas9为对照(Control)组;只转染pEGFP-C1-cas9质粒通过绿色荧光GFP的表达来检验转染是否成功。在荧光显微镜下观察发现,转染组的3D4/21细胞多数表达绿色荧光,表明质粒转染3D4/21细胞成功(图2)。
2.3 MiR-155在3D4/21细胞基因组中敲除效率的检测
收集转染后的3D4/21细胞提取基因组DNA后,以其作为模板进行PCR扩增,扩增包含miR155前体序列的696 bp的片段,PCR产物纯化试剂盒纯化以后,按照T7EⅠ核酸酶的说明书进行操作,用2%琼脂糖凝胶进行分析。随后用ImageJ定量软件计算切割效率,发现sgRNA51、sgRNA52、sgRNA42和sgRNA39均靶向敲除成功,流式分选前的sgRNA39、sgRNA42、sgRNA51、sgRNA52切割效率分别27%、32%、29%和28%,流式分选后分别为42%、41%、38%、36%(图3)。

表1 构建sgRNA表达载体引物、基因组扩增引物及荧光定量PCR引物信息
2.4 敲除miR-155的3D4/21细胞中缺失或插入位点测序验证
提取sgRNA39、sgRNA42、sgRNA51、sgRNA52实验组细胞的基因组DNA,经PCR扩增后连接19T载体,转化后挑单克隆送上海生工公司进行Sanger测序。分析测序结果发现,sgRNA39介导的Cas9核酸酶在靶点位置造成了31 bp、18 bp的缺失,sgRNA42介导的Cas9核酸酶在靶点位置造成了21 bp、11 bp的缺失,sgRNA51介导的Cas9核酸酶在靶点位置造成了24 bp、9 bp的缺失,sgRNA52介导的Cas9核酸酶在靶点位置造成了2 bp的缺失和插入 1 bp(图 4)。
2.5 敲除miR-155的3D4/21细胞中miR-155相对表达量的检测
为检测CRISPR/Cas9系统对3D4/21细胞内源miR-155的表达水平的影响,将转染pGL3-U6-sgRNA和pEGFP-C1-cas9质粒的细胞收集之后提RNA进行qPCR检测。qPCR结果显示,不管是miR-155-3p还是miR-155-5p,对照组都呈现高表达(对照组的表达量已标准化为1,图5)。与对照组相比,sgRNA39和sgRNA42实验组的miR-155-3p的表达量呈现极显著下调(P<0.01),sgRNA51的miR-155-3p的表达量呈现显著下调(P<0.05),sgRNA52的miR-155-3p的表达量没有下调,反而有少许增加;sgRNA39、sgRNA42、sgRNA51和 sgRNA52实验组的miR-155-5p的表达量均表现为显著下调(P<0.05)。不管是miR-155-3p还是miR-155-5p,sgRNA39实验组的表达量都是显著下调最明显的,暗示sgRNA39的敲除效率最高。
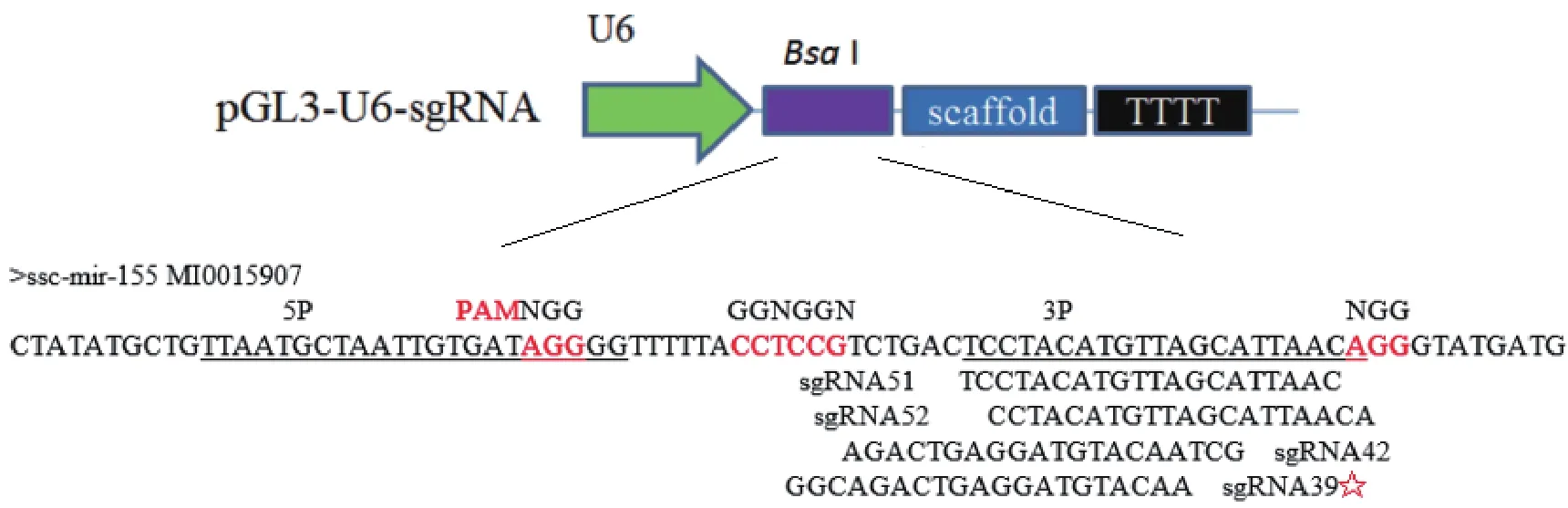
图1 构建靶向ssc-miR155的pGL3-U6-sgRNA载体
2.6 免疫印迹检测敲除miR-155的3D4/21细胞中靶基因表达水平
为了检测敲除miR-155的3D4/21细胞中,已验证的靶基因SHIP1的蛋白表达量,进一步验证miR-155的敲除效率,将转染pGL3-U6-sgRNA和pEGFPC1-cas9质粒的表达绿色荧光蛋白GFP的3D4/21细胞进行流式分选,富集表达GFP的细胞后提取蛋白样品,采用western blot方法进行miR-155靶基因SHIP1蛋白表达量的检测。结果(图6)显示,敲除miR-155的3D4/21细胞中,miR-155基因已验证的靶基因SHIP1呈现高表达,特别是敲除效率比较高的sgRNA39,实验组SHIP1表达量与对照组相比呈现明显上调表达(P<0.01)。

图2 pGL3-U6-sgRNA与pEGFP-C1-cas9载体共转染3D4/21细胞系(10×)
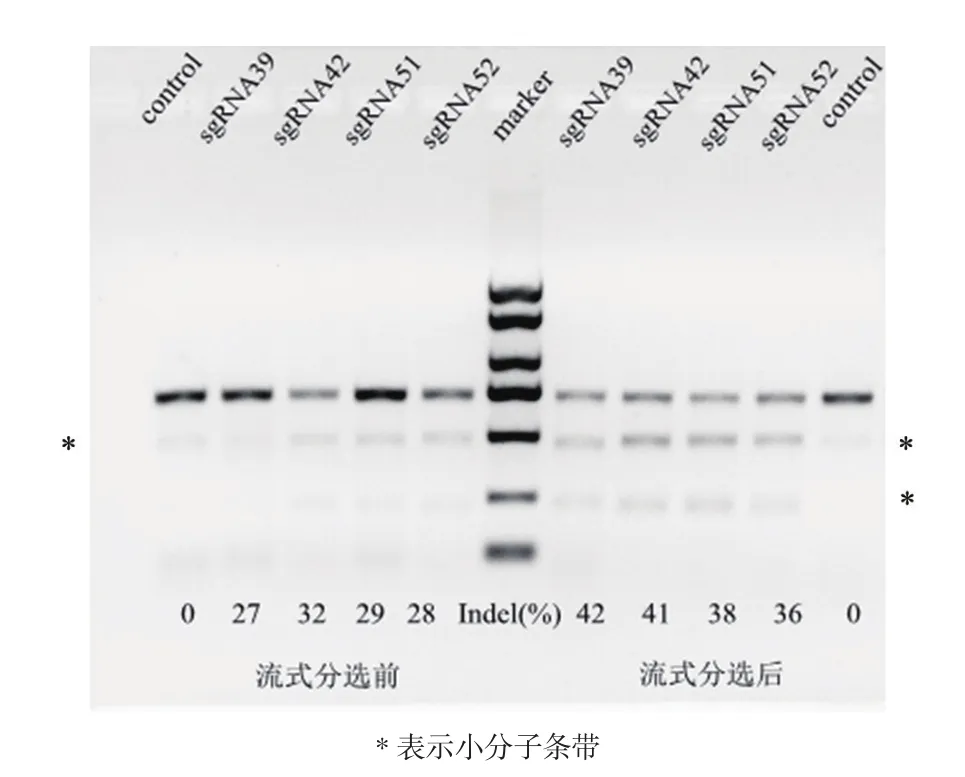
图3 T7EⅠ酶酶切检测sgRNA的切割效率

图4 Sanger测序结果
3 讨论
采用CRISPR/Cas9系统进行基因编辑,sgRNA的设计非常重要。Jing等[29]采用pLentiCRISPR系列慢病毒载体针对小鼠mmu-miR-155种子序列设计sgRNA构建表达载体,感染RAW264.7细胞后,建立单克隆细胞系,DNA测序结果显示 mmu-miR-155最多被敲除6 bp,qPCR结果显示mmu-miR-155的mRNA表达水平降低一半,正常培养条件下,靶蛋白SHIP1的表达量有少许上调,敲除效率不及本实验。最主要的原因可能是sgRNA引物的设计。本实验sgRNA是针对ssc-miR-155-3p序列设计的,所选4个sgRNA引物序列是评分较高的,其中sgRNA39评分最高,特别高于针对ssc-miR-155-5p序列设计的sgRNA;DNA测序显示sgRNA39敲除31 bp,qPCR结果显示sgRNA39敲除细胞中ssc-miR-155-3p表达量下调8倍多,ssc-miR-155-5p表达量下调10倍多;靶蛋白SHIP1的表达量上调8倍。
本实验中qPCR结果显示sgRNA52敲除细胞中ssc-miR-155-3p的表达量没有降低反而有少许上升,此反常现象可能的原因有:sgRNA52敲除效率是其中最低的,DNA测序结果显示有2 bp的敲除,或者是1 bp的插入。miR-155-3p/5p均参与真核基因表达调控,并且miR-155-3p/5p可通过两者表达量比值的改变来发挥调节功能[30]。本实验中sgRNA52敲除效率最低,而且测序结果显示有碱基插入或与参考序列一致,造成mRNA表达水平异常。
基因组编辑技术是研究基因功能的一个新型的重要工具。在CRISPR/Cas9技术出现之前,在细胞水平进行基因的功能研究主要采用RNAi技术。但RNAi属于瞬时转染,具有不彻底性,可能对基因功能的正确判断有影响,CRISPR/Cas9技术可以在基因组水平敲除目的基因,更适用于基因功能的研究[31]。本研究利用CRISPR/Cas9系统成功构建了miR-155敲除的3D4/21细胞株,解决了RNAi技术的瞬时性和不彻底性对基因功能研究的影响,在细胞水平上为更进一步深入研究miR-155的功能提供了更为有效的工具。
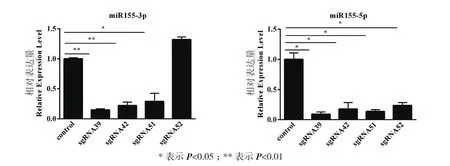
图5 qPCR检测miR155-3p和miR155-5p在不同敲除细胞中的表达水平
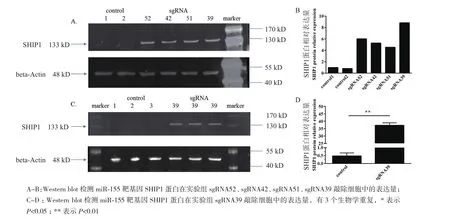
图6 Western blot验证miR-155靶基因SHIP1蛋白在敲除细胞中的表达量
4 结论
本研究采用带有GFP标记基因的CRISPR/Cas9系统,成功构建sgRNA表达载体,共转染3D4/21细胞,采用流式分选并富集表达GFP的细胞,收集细胞后分别在DNA、RNA和蛋白质水平检测敲除效率,结果显示T7EI酶酶切检测有小分子片段,Sanger测序显示在sgRNA附近位点有碱基敲除,qPCR检测miR-155-5p/3p表达量均有下降,western blot结果表明miR-155靶基因SHIP1的蛋白表达量升高。即成功构建miR-155敲除的3D4/21细胞系,为进一步探讨miR-155在巨噬细胞发挥的调控功能研究提供细胞模型。
猜你喜欢
北方牧业(2023年13期)2023-07-28 06:50:54
中老年保健(2021年7期)2021-08-22 07:40:46
今日农业(2021年11期)2021-08-13 08:53:24
无机化学学报(2020年7期)2020-07-20 02:06:44
三农资讯半月报(2020年8期)2020-05-13 14:26:35
柴油机设计与制造(2018年3期)2018-10-13 01:45:08
中国铸造装备与技术(2017年3期)2017-06-21 11:33:37
中国病理生理杂志(2017年2期)2017-01-17 03:59:14
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
