
应用CRISPR技术敲除MDA-MB-231细胞系的NODAL基因的研究
2019-11-21 11:09:30李念峰方天星倪玉芳曾凡才
生物技术通报 2019年11期
李念峰 方天星 倪玉芳 曾凡才
(西南医科大学基础医学院生物化学与分子生物学实验室,泸州 646000)
NODAL 是 TGF-β(Transforming growth factor-β)超家族成员之一,最初发现NODAL作为一个重要的形态发生素分子,主要在胚胎发育过程中发挥作用[1]。随着生物个体的生长发育,NODAL基因表达逐渐关闭,成年后仅发现在胎盘、子宫内膜和发育期乳腺等少数几种生殖相关的正常组织中表达。后有研究发现NODAL在黑色素瘤细胞中重新表达,又陆续发现它在乳腺癌、胶质瘤、前列腺癌、胰腺癌、子宫内膜癌、甲状腺癌和肝癌等肿瘤细胞中高表达,且可能与这些肿瘤的发生和发展相关[2-5]。综合文献分析和我们的研究经验发现,研究NODAL分子的功能具有一定的复杂性[3-6]。首先,在核酸和蛋白表达水平上准确检测NODAL表达存在一定的难度。最初发现NODAL存在促肿瘤作用的美国西北大学Hendrix MJ 实验室根据他们多年的研究经验提出在mRNA水平不易检测到NODAL基因的表达,分析多个实验室在核酸和蛋白水平检测NODAL表达的结果并不一致[7]。我们在研究过程中也面临同样的困惑。其次,TGF-β超家族不仅成员众多,而且这些细胞因子与受体存在一个配体可与多个受体结合,一个受体又可接受多个配体信号的特点[8]。TGF-β超家族至少有35个成员,它们属于细胞因子。传递这些细胞因子信号到胞内的受体属于丝氨酸/苏氨酸激酶受体,根据结构和功能不同分为Ⅱ型受体和I型受体,哺乳动物有5种II型受体和7种I型受体[9]。例如,研究发现NODAL分子至少可通过ALK4或ALK7两个I型受体传递信号,而ALK4和ALK7受体则还可接受包括NODAL分子在内的其它多种 TGF-β 超家族成员的信息[4,8,10-11]。最后,不同研究结果之间相互矛盾。多数研究认为NODAL基因具有促肿瘤作用,但有部分研究认为NODAL分子有抑制肿瘤的作用[12]。
研究基因功能的常用技术是基因敲除(Gene knock-out)或基因敲低(Gene knock-down)。已报道的文献均采用的基因敲低NODAL表达后观察对肿瘤细胞功能的影响。由于基因敲低技术一般不容易将基因表达完全抑制,另一方面这种抑制作用对基因表达的影响也是暂时。鉴于NODAL基因研究的复杂性,本研究拟通过基因敲除以及恢复该基因表达以进一步研究NODAL基因在肿瘤细胞中的功能。并且还可通过敲除NODAL基因验证NODAL抗体的特异性。
乳腺癌在中国女性中发病率最高,死亡率位居所有癌症的第6位[13]。其中三阴性乳腺癌的预后最差,因此本研究选取三阴性乳腺癌细胞株MDAMB-231作为基因敲除的靶细胞,采用CRISPR/Cas9技术敲除该细胞内NODAL基因,为进一步揭示NODAL基因在乳腺癌细胞中的分子机制奠定基础。
1 材料与方法
1.1 材料
1.1.1 主要试剂 MDA-MB-231细胞株、UCATMCRISPR/Cas9快速构建及活性检测试剂盒、AIOTMgene KO Cell Line Kit(ProuR)、CRISPR 质 粒 均 购自北京百奥赛图基因生物技术有限公司。嘌呤霉素(Puromycin)购自北京索莱宝科技有限公司。质粒提取试剂盒、DNA纯化试剂盒、琼脂糖凝胶回收试剂盒购自广州Omega飞扬生物工程有限公司。限制性核酸内切酶、T4连接酶购自宝日医生物技术(北京)有限公司,细胞培养相关试剂购自美国Thermo公司。
1.1.2 主要实验仪器 细胞培养箱、PCR仪、凝胶成像仪、恒温摇床、恒温培养箱、荧光显微镜、生物安全工作台。
1.2 方法
1.2.1 敲除方案及Cas9/sgRNA质粒构建 利用NCBI(https://www.ncbi.nlm.nih.gov/) 查 找NODAL基因信息,分析NODAL基因生物学特征,根据NODAL基因CDS区结构及性质,确定敲除位点,制定敲除NODAL基因第二外显子的敲除方案(图1)。以MDA-MB-231细胞系cDNA为模板,设计引物并用PCR扩增靶位点序列,测序确认靶序列与GenBank中的NODAL基因参考序列一致,并利用此序列信息设计sgRNA,以确保Cas9/sgRNA的效率。
利用CRISPR在线设计工具(http://CRISPR.mit.edu/),在NODAL基因第二外显子位置5'端设计了7条sgRNA序列,即sgRNA1-7,3'端设计了4条sgRNA序列,即sgRNA8-11(表1),由英潍捷基(上海)贸易有限公司合成sgRNA oligo及其互补链。
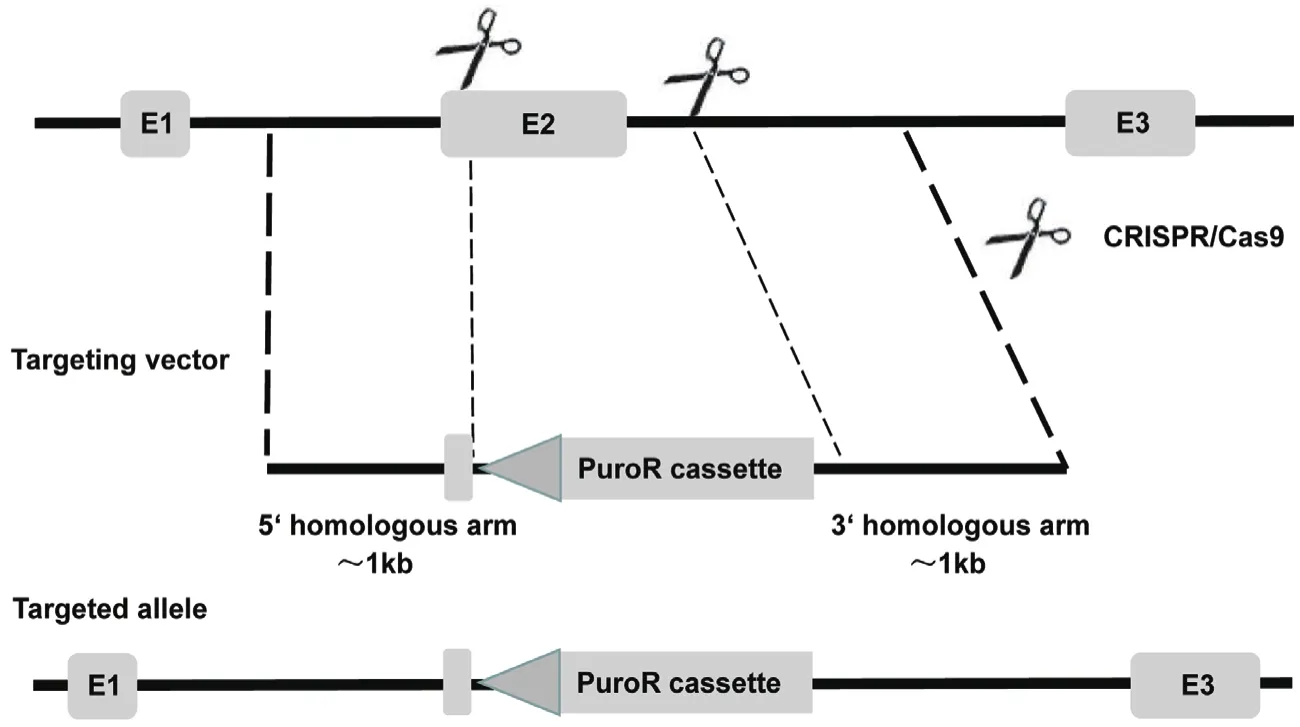
图1 敲除方案示意图

表1 sgRNA oligo设计
sgRNA oligo经变性-退火之后形成sgRNA oligo双链,利用UCATMCRISPR/Cas9快速构建及活性检测试剂盒连入BbsI核酸内切酶处理后的PCS质粒载体中,通过测序验证,确定构建成功后,分别命名为PCS-sgRNA1-PCS-sgRNA11。利用UCATMCRISPR/Cas9快速构建及活性检测试剂盒检测PCS-sgRNA活性,经数据分析,在5'端和3'端选择最佳PCS-sgRNA质粒用于下一步实验。
1.2.2 打靶载体构建 基于同源重组修复机制敲除目标基因的原理,打靶载体由位于靶位点上游的左同源臂(L-arm,LR)和下游的右同源臂(R-arm,
RR)以及两者之间的嘌呤霉素抗性基因PuroR构成。因此,分别在NODAL基因第二外显子上游和下游设计引物克隆同源臂,设计克隆左同源臂的PCR引 物 F:5'-CGATGGTACCGAAGCTTCTGGGTGCA TGTGATTGC-3',下划线处为KpnⅠ酶切位点,R:5'-CGATGTCGACAGAGGTTGGAGTAGAGCATAAGG AGC-3',下划线处为SalⅠ酶切位点,扩增产物片段大小为1285 bp;右同源臂PCR引物F:5'-CGATA CGCGTTTGGAATCACACTGTTCCAGCCACAG-3',下划线处为MluⅠ酶切位点,R:5'-CGATGCGGCCGC CTACCTTTAGCTCTGTGCTTGTTTTGTG-3',下划线处为NotⅠ酶切位点,扩增产物片段大小为1491 bp。上述4条引物中5' CGAT均为添加的酶切保护碱基。采用相应引物PCR扩增,经凝胶电泳鉴定后胶回收,利用AIOTMgene KO Cell Line Kit试剂盒将LR、RR、PuroR经一步克隆一起连入LScKO-4G质粒中,构建LScKO-LR-RR质粒。再利用HindⅢ+SalⅠ、XhoⅠ+BamHⅠ、EcoRⅠ+NotⅠ等限制性核酸内切酶组合酶切鉴定打靶载体,最终通过测序确认是否构建成功。
1.2.3 电转及筛选 以1350 V电压将作用于5'端和3'端的PCS-sgRNA质粒以及打靶载体质粒按质量比1∶1∶1转染入MDA-MB-231细胞中。无CO2条件下培养至细胞达到80%及以上融合度时,加入2μg/mL嘌呤霉素进行药物筛选,直至无大量细胞死亡时,取部分细胞提取基因组DNA,然后进行混合基因型PCR鉴定。
1.2.4 混合基因型PCR鉴定及单细胞克隆挑选 如果电转后在靶位点发生了同源重组,打靶载体提供的外源PuroR基因DNA会与靶位点DNA整合。因此,可在左同源臂上设计一条引物HR-F:5'-TGGTTGTCTTGGGTGTGTGGAACAG-3',在PuroR外源基因靠近左同源臂端设计一条引物HR-P-F:5'-CTCGACTGTGCCTTCTAGTTGCCAG-3',扩增产物片段大小为1782 bp。同理,在右同源臂上设计一条引物HR-R:5'-CAGAGGCCACTTGTGTAGCG-3',在PuroR外源基因靠近右同源臂端设计一条引物HRP-R:5'-CAATTCACATCCTAGATGGGCAGGTC-3',扩增产物片段大小为1780 bp(图2)。取部分经电转且嘌呤霉素筛选后存活的细胞,提取基因组DNA作为模板,如果这两对引物能够扩增出预期大小的条带,说明电转后有细胞发生了同源重组,可进行单细胞克隆挑选。
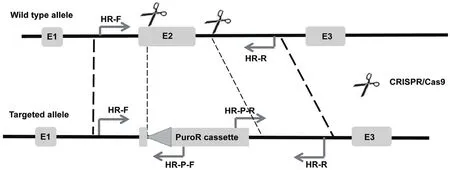
图2 等位基因同源重组PCR基因型鉴定引物位置示意图
1.2.5 单细胞克隆基因型鉴定
1.2.5.1 等位基因同源重组PCR鉴定 等位基因同源重组PCR鉴定同步骤1.2.4,差别仅在于此处是鉴定单细胞克隆的基因型(图2)。
1.2.5.2 等位基因移码突变PCR鉴定 本研究共设计两个Cas9/sgRNA酶切作用位点,在靠近左同源臂的作用位点两端设计一对引物FS-F:5'-GCTGCTTAGAGCGGTTTCAGATGGA-3'和FS-R:5'-GTCGGATGAAACTCCTCCCCAACAG-3',野生型基因扩增产物片段大小为493 bp,如果发生了移码突变,扩增产物片段大小非常相似,只能通过测序判断基因型(图3)。
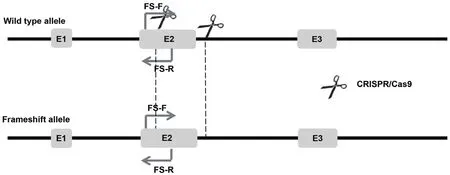
图3 等位基因移码突变PCR鉴定引物位置示意图
1.2.5.3 等位基因大片段缺失PCR鉴定 在设计的两个Cas9/sgRNA酶切作用位点的两端,设计一对引物,Del-F:5'-TTGTCCCAGGTCACCTTTTCCTTGG-3'和 Del-R:5'-TTCTTCCTCCACCACCTCCTAGACC-3',野生型基因扩增产物片段大小为1462 bp,发生同源重组的基因扩增产物大小为3151 bp,如果是发生了大片段缺失的片段大小约为620 bp(图4)。最终通过测序确定单细胞克隆是否发生等位基因大片段缺失。
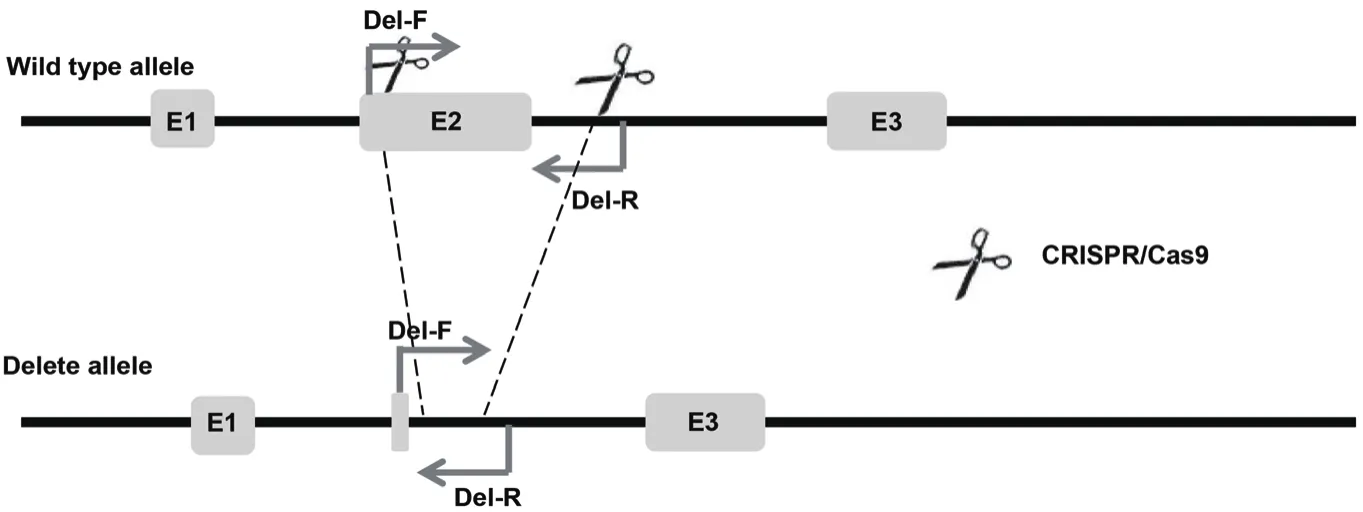
图4 等位基因大片段缺失PCR鉴定引物位置示意图
1.2.6 NODAL蛋白水平鉴定 将基因型鉴定为阳性的单细胞克隆扩大培养,提取细胞总蛋白,经SDSPAGE电泳并转至PVDF膜,用5%脱脂奶粉封闭1 h,分别用NODAL一抗(Abcam,Cat# ab55676,1∶1000)和 β-actin一抗(1∶10000)4℃孵育过夜,1×TBST清洗后,用山羊抗鼠(1∶5000)、山羊抗兔(1∶5000)二抗溶液中室温孵育1 h,1×TBST再次清洗后,用化学发光仪检测NODAL蛋白表达情况。
1.2.7 统计分析 Cas9/sgRNA质粒的活性检测,利用SPSS 19.0软件将五次重复所得到的数据通过方差检验计算每组的平均值±标准误差,并以Con组为1作为标准,计算各组相对sgRNA活性。
2 结果
2.1 Cas9/sgRNA质粒构建及活性鉴定
基于sgRNA的设计原则,5'端设计了7条sgRNA,3'端设计了4条sgRNA,分别与CRISPR载体质粒PCS连接,经测序确认成功构建11个作用于NODAL基因靶序列的 Cas9/sgRNA质粒。使用SSA reporter assay检测sgRNA活性,各PCS-sgRNA质粒显示出不同程度的活性。根据质粒活性并结合sgRNA设计网站显示这几条sgRNA的不同特异性等因素综合考虑,最终5'端选择PCS-sgRNA1(图5-A),3'端选择PCS-sgRNA8(图5-B)两种Cas9/sgRNA质粒进行后续基因敲除实验。
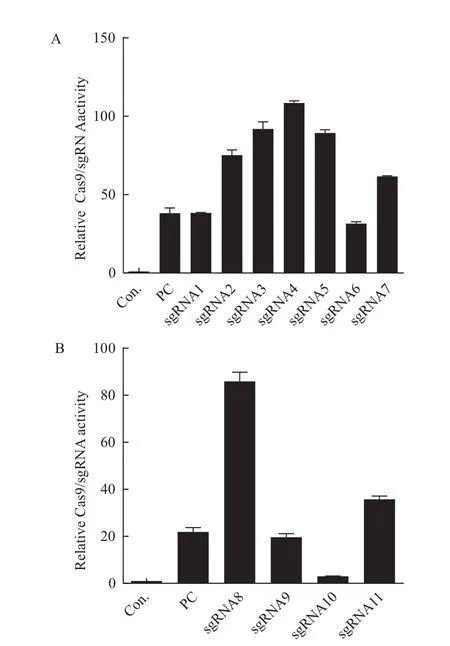
图5 Cas9/sgRNA质粒活性检测
2.2 打靶载体构建
以MDA-MB-231乳腺癌细胞基因组DNA为模板,用设计的PCR引物分别扩增左、右同源臂,电泳结果(图6-A)显示片段大小与预期相符。将左、右同源臂与试剂盒中的PuroR基因一起克隆至LScKO-4G质粒中,构建LScKO-LR-RR质粒,然后使用限制性核酸内切酶酶切验证打靶载体。根据实验设计,如果用Hind Ⅲ+SalⅠ双酶切打靶载体质粒,电泳后应观察到3条带,大小应分别为1264 bp、2727 bp和4059 bp;如果用XhoⅠ+BamHⅠ双酶切打靶载体质粒,电泳后也应观察到3条带,条带大小分别为386 bp、2228 bp和5436 bp;而如果用EcoRⅠ+NotⅠ双酶切打靶载体质粒,电泳后也应观察到3条带,条带大小分别为768 bp、2754 bp和4528 bp。应用这3种组合酶酶切打靶质粒的琼脂糖凝胶电泳结果显示片段大小与预期一致(图6-B)。只是XhoⅠ+BamHⅠ双酶切后产生的386 bp和EcoRⅠ+NotⅠ双酶切产生的768 bp两条带均由于DNA片段小,结合EB染料少,观察到的条带弱,在本图中未能清晰展示(图6-B)。最终经测序确认,打靶载体构建成功。
2.3 基因型鉴定
2.3.1 混合基因型鉴定 以电转并经嘌呤霉素筛选后存活的部分细胞提取基因组DNA作为模板,采用鉴定基因重组的引物扩增DNA片段。根据实验设计,如果外源的PuroR基因与NODAL基因第二外显子靶位点发生重组,利用外源基因5'端位置附近设计的PCR引物扩增的产物大小为1782 bp,外源基因3'端位置附近设计的PCR引物扩增的产物大小为1780 bp。这两对引物的PCR产物琼脂糖凝胶电泳结果与预期大小均一致(图7),说明至少在部分细胞中PuroR基因与NODAL基因靶位点发生了同源重组,可挑取单细胞克隆进行后续实验。
2.3.2 等位基因同源重组基因型鉴定 单细胞克隆等位基因同源重组基因型鉴定与混合基因型鉴定原理一样,不同之处在于本鉴定是以单细胞克隆中提取的DNA作为PCR扩增的模板。PCR产物预期大小也是5'端为1782 bp,3'端为1780 bp。将挑取的15个单细胞克隆的基因组DNA作为模板,PCR产物电泳结果显示,在15个单细胞克隆中,仅有C1单细胞克隆的5'端PCR产物条带较弱,其余单细胞克隆条带强,且与预期大小相符(图8-A);而在15个单细胞克隆的3'端PCR产物中,C1单细胞克隆未观察到与预期大小一致的条带,B6单细胞克隆的目标条带较弱,其余单细胞克隆均有与较强的目标条带(图8-B)。最终选择A4、B4、B6、C2、E4、F3、F4和H3共8个单细胞克隆的PCR产物进行测序,测序结果显示这8个单细胞克隆的NODAL等位基因均发生了同源重组。
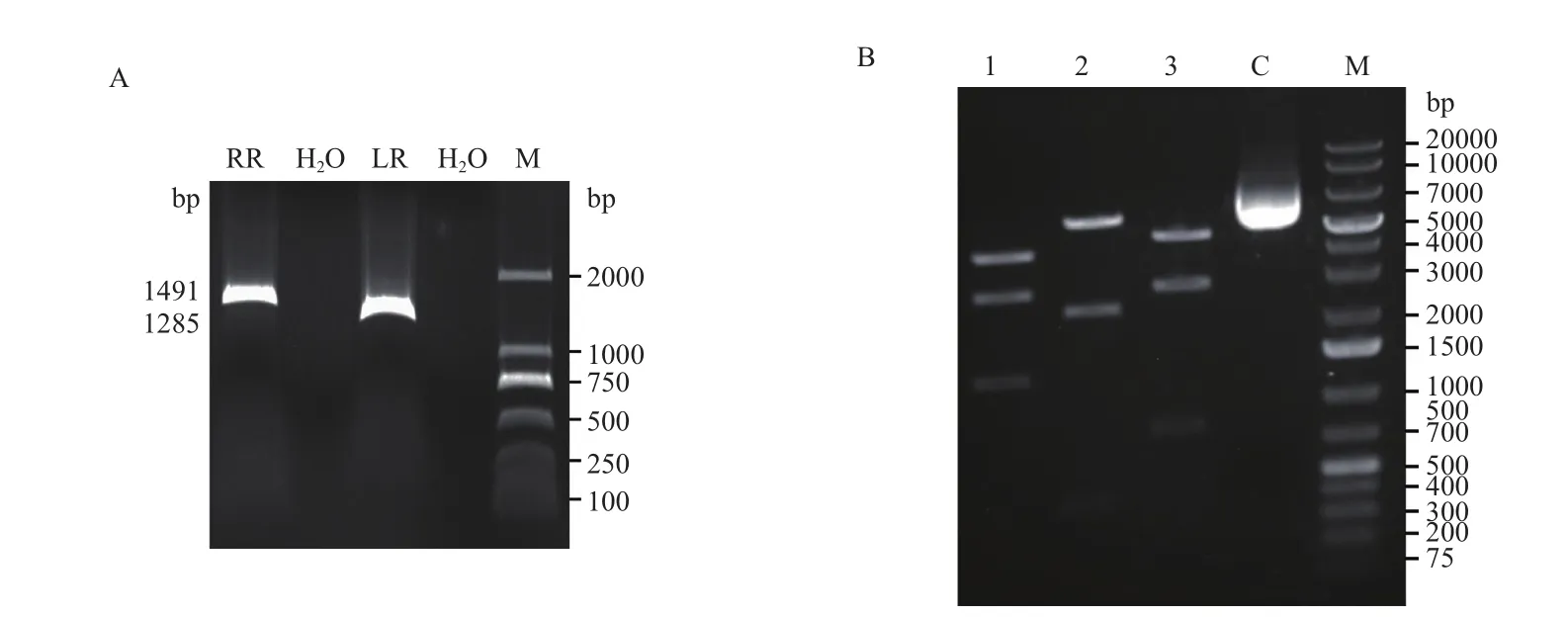
图6 打靶载体构建
2.3.3 等位基因移码突变基因型鉴定 通过等位基因同源重组基因型鉴定阳性的单细胞克隆,仅能证明至少有一个NODAL基因的等位基因发生了重组替换。但是癌细胞至少有2个以上的等位基因。此外,当Cas9/sgRNA复合体在靶位点剪断DNA双链后,细胞除可利用同源重组的修复机制外,还可利用非同源末端连接的修复机制修复DNA损伤,从而造成靶基因发生移码突变,同样可达到敲除基因的效果。因此,需进行移码突变基因型进行鉴定。利用扩增等位基因移码突变的引物进行PCR扩增,结果显示:15个单细胞克隆中,除B4、C2、F4三个单细胞克隆外,其余12个单细胞克隆均扩增出大小约为490 bp的条带(图9-A),将其中A4、B6、E4、F3和H3五个单细胞克隆的PCR产物进行了全长测序。测序结果经比对分析,发现B6、E4、F3和H3四个单细胞克隆扩增产物是野生型片段(扩增序列与NODAL基因参考序列完全一致,比对结果未展示),说明在这4个细胞克隆中,至少还有一个野生型NODAL基因拷贝。仅有A4单细胞克隆在NODAL基因mRNA序列的642位插入了一个T碱基(图9-B),该插入T碱基位于PCR产物正向测序峰图的191位,且峰形完整(图9-C),反向测序峰图在相应位置也有完整峰形的该插入T碱基(结果未展示)。上述结果表明,A4单细胞克隆在第二外显子区域插入非三的整倍体碱基,发生等位基因移码突变。
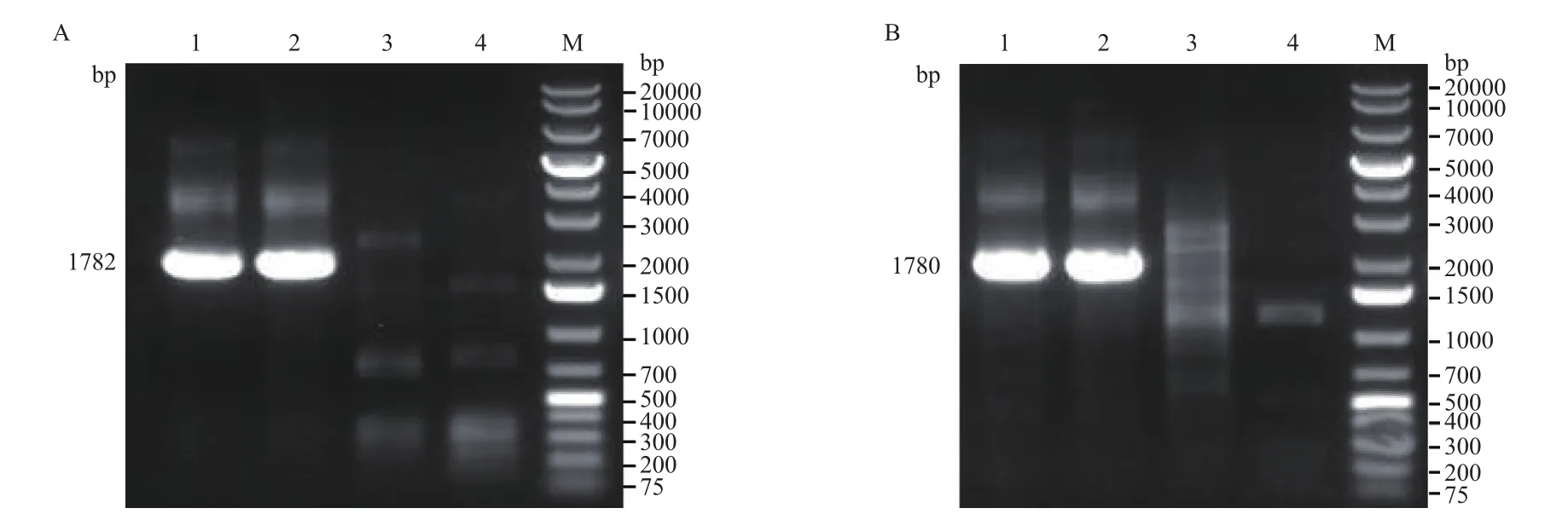
图7 混合克隆细胞基因型鉴定
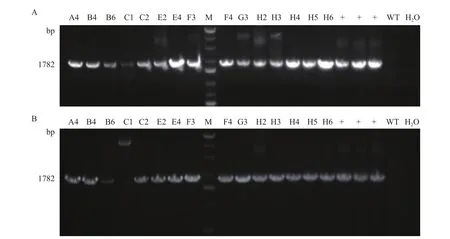
图8 等位基因同源重组PCR鉴定
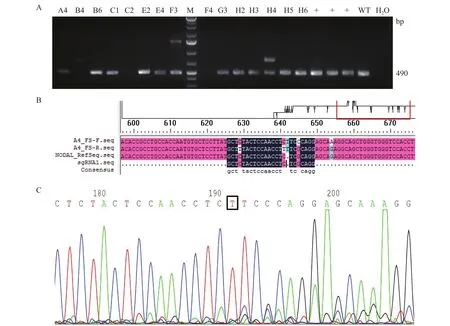
图9 等位基因移码突变基因型鉴定
2.3.4 等位基因大片段缺失基因型鉴定 由于本研究采用双位点剪切靶序列,还可能发生基因大片段缺失的情况。利用检测大片段缺失的引物进行PCR扩增。结果(图10-A)显示,8个单细胞克隆仅E4、F3、F4和H3单细胞克隆能扩增出的大小约为620 bp的条带,说明这4个单细胞克隆可能发生了等位基因大片段缺失。将F3约620 bp的PCR产物克隆并测序的结果进行基因组BLAST比对,结果(图10-B)显示在10号染色体上NODAL基因位置从编号70434736 bp到70435576 bp部分发生DNA片段缺失,大小为840 bp。同样方法还分析了E4、F4和H3单细胞克隆的测序结果,结果与F3类似,其中E4缺失了848 bp基因组片段,F4缺失了840 bp基因组片段,H3缺失了840 bp基因组片段(基因组BLAST比对结果未展示)。
综合分析等位基因同源重组、等位基因移码突变和等位基因大片段缺失这3种PCR基因型鉴定及测序结果,总结为表2。结果显示;A4,B4,C2 和F4四个单细胞克隆的NODAL基因均被敲除,B6,E4,F3和H3四个单细胞克隆的NODAL基因被部分敲除。
2.4 蛋白表达鉴定
将已进行基因型鉴定的单细胞克隆全部扩大培养,提取总蛋白进行了NODAL蛋白表达检测。结果显示在8个单细胞克隆中,检测到B4、E4、F3三个单细胞克隆的NODAL蛋白表达水平有不同程度降低,A4、B6两个单细胞克隆检测不到NODAL蛋白的表达,其他3个单细胞克隆NODAL蛋白表达变化不大(图11)。说明A4、B6在蛋白水平鉴定为敲除NODAL基因的单细胞克隆。
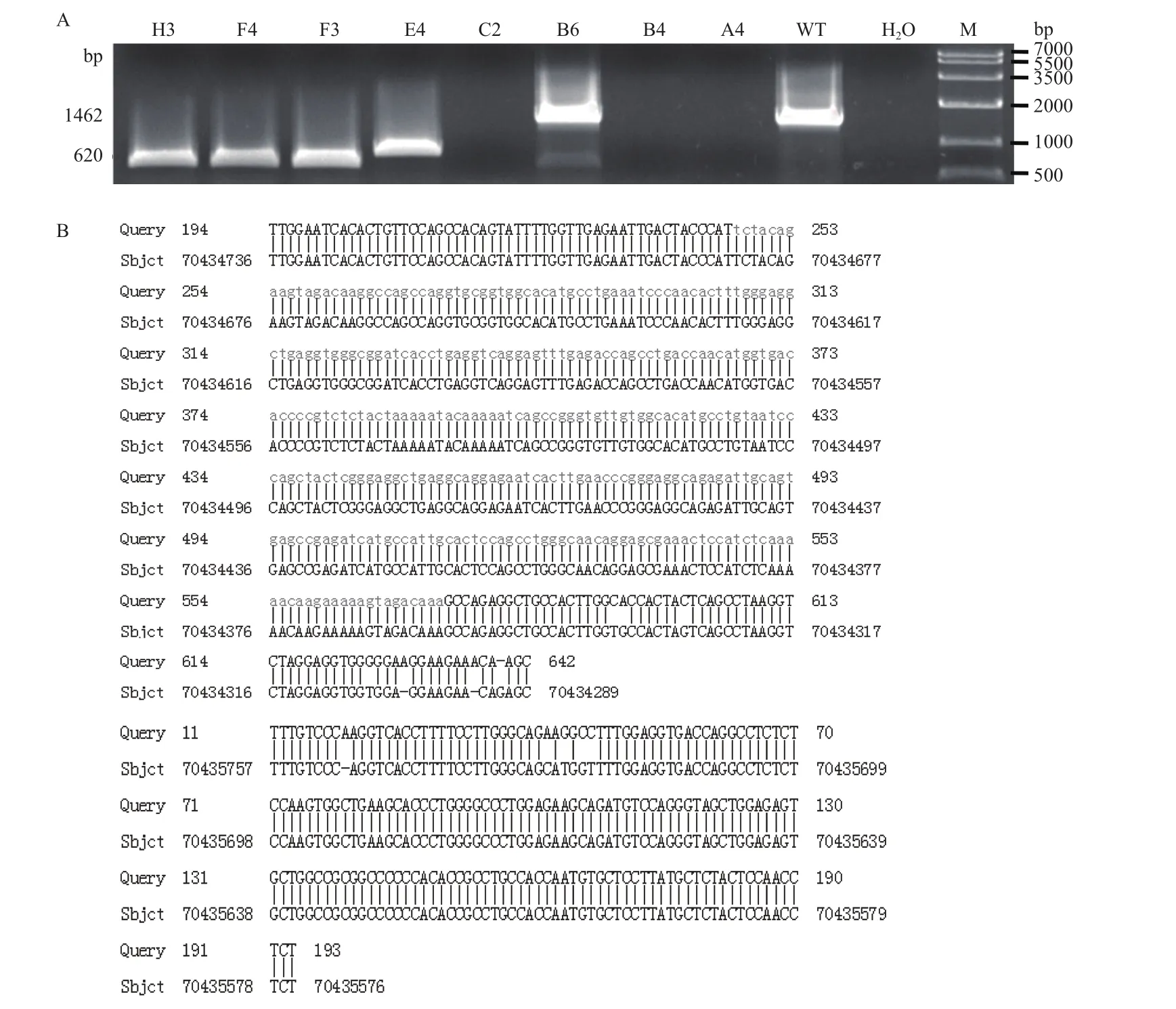
图10 等位基因大片段缺失基因型鉴定
3 讨论
自1993年克隆NODAL基因以来,发现该基因主要在胚胎发育过程中有重要作用[14]。至2006年Hendrix实验室发现NODAL在黑色素瘤中有促肿瘤作用,后续研究发现在其它多种肿瘤中也有同样作用[15-16]。检索到关于NODAL在肿瘤细胞中功能的文献均采用基因敲低技术抑制NDOAL的表达,未见敲除NDOAL基因细胞系的报道。本研究利用CRISPR/Cas9基因编辑技术在三阴性乳腺癌细胞MDA-MB-231中成功敲除NODAL基因。
CRISPR/Cas9基因编辑技术是继ZFNs、TALENs技术之后的第三代基因编辑技术。该技术是在向导RNA的指导下,核酸内切酶Cas9蛋白可将靶DNA在特定位点剪断,造成DNA双链断裂的损伤,再借助细胞自身具有的非同源末端连接和同源重组修复的机制,引发剪切位点的DNA碱基插入或缺失以及DNA片段的替换,最终使靶基因被敲除[17]。为了提高敲除效率,本研究提供了有同源片段的打靶载体,通过同源重组机制达到敲除基因的目的。本实验利用CRISPR/Cas9基因编辑技术精确剪切MDAMB-231乳腺癌细胞系NODAL基因第二外显子5'端和3'端处两个位点后,再通过同源重组机制将外源嘌呤霉素抗性基因替换NODAL基因的Exon2,从而达到敲除NODAL基因的目的。CRISPR/Cas9基因编辑技术具有操作简单、实验周期短、成本较低等优点,广泛用于各种生物学、医学研究[18]。
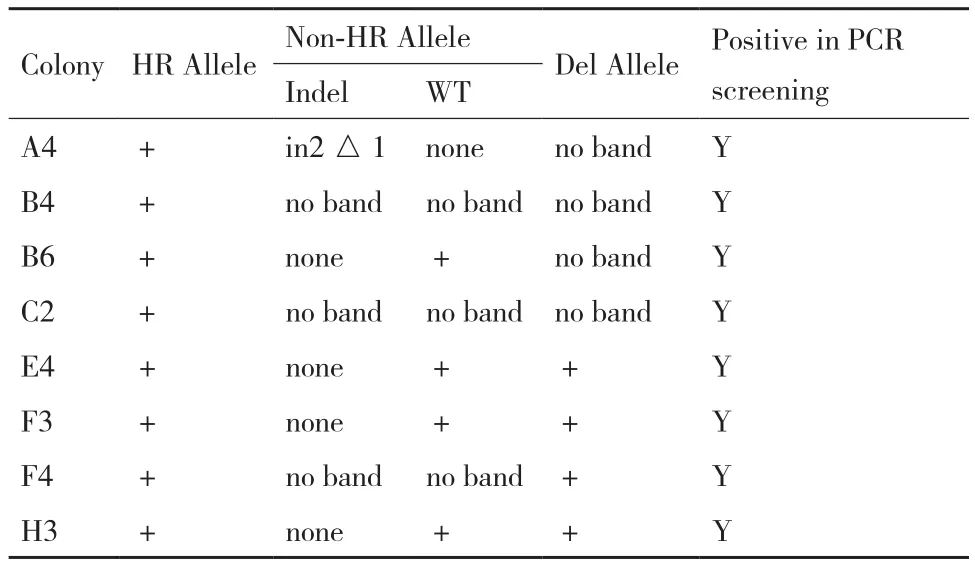
表2 基因型鉴定汇总
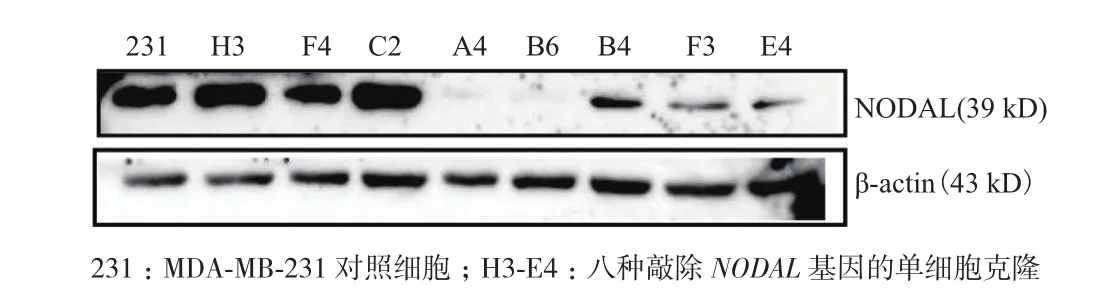
图11 Western blot检测NODAL蛋白表达
NODAL分子除TGF-β超家族成员所具有的信号转导复杂性外,它还具有在mRNA和蛋白表达水平检测的复杂性。mRNA表达水平不易检测,存在较复杂的剪接异构体,以及反义转录本和环状转录本[19-20]。而NODAL蛋白表达虽然容易检测,但是不同文献报道的蛋白质分子量差异较大[7]。已报道的NODAL前体蛋白的分子量范围为35-48 kD,成熟肽的分子量范围是13-17 kD,且不易在细胞裂解液中检测到NODAL成熟肽。我们在研究过程中也同样面临NODAL分子在mRNA和蛋白水平检测的问题,以至于我们对NODAL抗体的特异性持怀疑态度,先后购买了5种NODAL抗体进行检测,检测结果差别较大。由于基因敲除是验证抗体特异性的好方法,通过本研究对NODAL基因的敲除和验证,发现仅有Abcam(ab55676)和Santa Cruz(W65)两种抗体的结果更可信。
鉴于NODAL研究的复杂性,本研究从等位基因的同源重组、移码突变和大片段缺失3个方面鉴定单克隆细胞的基因型,以便于在基因组水平分析靶细胞的NODAL基因被敲除情况。结果发现B6单细胞克隆虽然还存在野生型的NODAL基因拷贝,但是却检测到NODAL蛋白不表达或很低表达,这说明即使有残留NODAL基因拷贝但表达效率已很低。由于癌细胞的染色体是非整倍体,基因存在多个拷贝,完全敲除癌细胞的基因有一定难度。如果经一轮敲除仅获得部分基因拷贝敲除的细胞,可将此细胞再进行一轮敲除,可提高成功率。本研究还发现虽然有的单细胞克隆鉴定为NODAL基因被部分或完全敲除,但是却未影响NODAL蛋白的表达,甚至有蛋白表达增加的趋势。这些问题还有待进一步探究。此外,后续我们还将利用这些敲除细胞进行体外和体内实验,以研究NODAL基因在乳腺癌细胞增殖、迁移、侵袭、转移和代谢等方面的作用。
4 结论
应用CRISPR/Cas9基因编辑技术并利用DNA断裂损伤后同源重组和非同源重组修复的机制,最终获得基因型和蛋白表达鉴定为NODAL基因被敲除的单细胞克隆,为进一步研究NODAL分子在乳腺癌细胞中的功能提供了重要支撑。本研究还发现有一些单细胞克隆虽然基因型鉴定为阳性,但是NODAL蛋白的表达并不降低,其原因还有待进一步研究。
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
华人时刊(2023年1期)2023-03-14 06:43:36
汉字汉语研究(2021年2期)2021-08-30 08:58:46
智慧健康(2021年17期)2021-07-30 14:38:32
科学(2020年4期)2020-11-26 08:27:16
现代检验医学杂志(2016年5期)2016-08-20 03:16:54
河北书画研究(2016年3期)2016-04-28 08:55:35
法医学杂志(2015年4期)2016-01-06 12:36:40
生殖医学杂志(2015年11期)2015-02-28 16:32:12
化学工业与工程(2015年1期)2015-02-10 03:01:41
