
鼠冠状病毒核衣壳蛋白电荷非均一性与其磷酸化的相关性
2019-10-29 03:41文荣王玉燕叶荣
微生物与感染 2019年5期
文荣,王玉燕,叶荣
复旦大学上海医学院基础医学院,教育部、卫健委、医科院医学分子病毒学重点实验室;病原生物学系,上海 200032
冠状病毒(coronaviruses)是一类有包膜的单股正链RNA病毒,感染人或动物后引起呼吸道、消化道及中枢神经系统病变[1]。冠状病毒基因组为目前发现的最大mRNA,长度为26.4~31.7 kb[1]。冠状病毒属于套式病毒目(Nidovirale),复制过程中形成一系列有着共同5′-前导序列和3′-下游序列的亚基因mRNA,用于表达病毒不同的蛋白质,这些较长mRNA的保真度依赖于该类病毒复制酶中外切酶ExoN的活性[2]。鼠肝炎病毒(mouse hepatitis virus, MHV)作为乙型冠状病毒属(betacoronavirus)的原型种,迄今为止仍是冠状病毒分子机制和致病性研究的理想模型[3-4]。
病毒的核衣壳(Nucleocapsid, N)蛋白结构相对保守,具有较强的同源和异源蛋白相互作用的能力,在病毒复制过程中能特异性识别并结合病毒的mRNA进而调控病毒复制,最终与基因组结合构成病毒的核心[5]。鼠冠状病毒MHV-A59的N蛋白含454残基,构成两个保守结构域, 即N端结构域(N-terminal domain,NTD)和C端结构域(C-terminal domain,CTD)。NTD的结构及电荷分布与其他冠状病毒如严重急性呼吸综合征冠状病毒(severe acute respiratory syndromes coronavirus,SARS-CoV)、HCoV-OC43和IBV高度一致[6]。已有实验证明磷酸激酶GSK-3通过催化SARS-CoV N蛋白S177和鼠冠状病毒MHV-JHM N蛋白S197磷酸化,促进了N蛋白招募RNA解旋酶DDX1,从而提高模板切换来合成更长的亚基因组mRNA[7-8]。
蛋白激酶C(protein kinase C, PKC)是重要的细胞信号转导中介分子,参与细胞的应激增殖分化、凋亡等[9]。PKC有9种同工酶,属于AGC亚类,根据调节结构域和激活方式不同进一步分为常规、新型和非典型3个亚组,其活化依赖于小分子的结合、膜转位和3个位点的磷酸化[10]。实验证明PKC与多种病毒的复制和致病作用密切相关,如博尔纳病病毒(Borna disease virus, BDV)感染导致各种神经系统症状,是由于病毒聚合酶辅助因子P蛋白通过PKC磷酸化并劫持其参与的神经元重要信号通路分子所致[11];PKC拮抗剂具有激活潜伏的人类免疫缺陷病毒1型(human immunodeficienty virus type 1, HIV-1)且能降低CD4+T细胞受体表达的双重作用,可提高高效抗反转录病毒疗法(highly active antiretroviral therapy, HAART) 的效果[12];另外,最近发现PKC通过结合流感病毒PB2招募磷酸化核衣壳蛋白单体聚集,从而与RNA形成有效的复制复合物[13]。
我们用鼠冠状病毒MHV-A59感染的小鼠神经母细胞瘤细胞系neuro-2a,通过细胞蛋白脂酰化抑制剂2-溴棕榈酸(2-bromopalmitate, 2-BP)和蛋白酶体抑制剂MG-132处理,结合2D电泳和免疫印迹试验分析了病毒复制及其N蛋白电负性与磷酸化之间的关系。结果提示鼠冠状病毒复制及其N蛋白磷酸化不但与PKCα的活化水平有关,并且某些内质网相关蛋白降解(endoplasmic reticulum-associated protein degradation, ERAD)与相关蛋白组分折叠也参与了这一过程。
1 材料和方法
1.1 细胞及培养
neuro-2a细胞(ATCC® CCL-131)为小鼠脑神经母细胞瘤细胞系[14],购自中科院细胞生物学研究所;L2细胞(ATCC® CCL-149)为大鼠肺上皮细胞系,来自美国纽约州卫生署Paul S. Masters博士实验室[15]。两种细胞均用含10%胎牛血清(Gibco)的高糖DMEM(HyClone)培养基,于37 ℃、5%CO2环境(CONTHERM MITRE SERIES 4000培养箱)中培养。细胞用1∶2~3稀释的0.25%胰酶-EDTA(HyClone)消化,按1∶3~5培养瓶面积比分瓶,间隔3~4 d,俟细胞汇合度接近80%时传代。小分子抑制剂处理鼠冠状病毒感染细胞采用含2%胎牛血清的VP-SFM(Gibco)培养基(2%FBS/VP-SFM)。
1.2 病毒感染与滴度测定
鼠冠状病毒MHV-A59野生株源自Paul S. Masters实验室分离株Alb240[15]。病毒扩增取高滴度贮存液1 ml〔(1~5)×107PFU/mL〕,感染1个T25 neuro-2a细胞1 h,补完全培养基4 ml后培养48 h,至CPE≥80%时收集5 ml培养上清液,连续感染1个T75 neuro-2a细胞1 h,补无血清培养基20 ml培养48 h后收集上清液25 ml,离心除去细胞碎片后分装并低温储存,同时测定病毒滴度。含高滴度病毒培养上清液合并后也可用于病毒颗粒纯化,初步纯化通常采用2次PEG沉淀[16]。
小分子抑制剂处理鼠冠状病毒感染细胞:2-BP(Sigma)和MG-132(Calbiochem)分别用无水乙醇(国药集团)和DMSO(Sigma)配成10 mmol/L储存液,临用前取适量用2% FBS/VP-SFM稀释至终浓度,分别为25 μmol/L和10 μmol/L。鼠冠状病毒储存液根据培养皿和细胞数用2% FBS/VP-SFM稀释至5×105PFU/mL(通常1.0 MOI),感染neuro-2a细胞1 h后吸去,换用含抑制剂的培养基继续培养,分别在病毒感染后12、24和48 h收集培养上清液和细胞裂解液样品。
病毒滴度用L2细胞噬斑法测定。L2细胞用D60培养皿培养36~48 h,直至达70%~80%汇合度,吸尽细胞培养基。将病毒上清液用含2%血清的DMEM按10倍系列稀释,取合适稀释度病毒1 mL加至细胞。37 ℃ 感染1 h后吸尽病毒稀释液,加入7 mL 55 ℃温育的琼脂细胞培养液(0.95%琼脂/5% FBS/DMEM),凝固后在37 ℃ 、5%CO2条件下继续培养约40 h,再覆盖3 mL 55 ℃ 温育的含0.05%中性红的琼脂细胞培养液,37 ℃ 、5%CO2条件下染色约8 h,计数噬斑数量。
1.3 N基因克隆与原核表达
MHV-A59 N基因克隆与原核表达采用已发表的方法[16],即以质粒pMH54[15]为模板,通过聚合酶链反应(polymerase chain reaction,PCR)获得鼠冠状病毒N蛋白基因片段(mN)。正向引物mN-1F序列为 5′-CGCCATATGATGTCTTTTGTTCC-TGGG-3′,反向引物mF-2R序列为5′-CGCAA-GCTTTTACACATTAGAGTCATCTT-3′。PCR片段用NdeⅠ和HindⅢ 位点克隆至载体pET-28a中6-His标签下游,转化大肠埃希菌感受态细胞DH5α。含目的基因质粒pET-28a-m阳性克隆用PCR筛选和测序确认,然后制备小量质粒并保存。重组MHV-A59 N蛋白表达首先用pET-28-mN转化大肠埃希菌感受态细胞BL21,然后挑取单个菌落接种于100 mL含50 μg/mL卡那霉素的LB培养基中,25 ℃ 下220 rpm摇菌至OD600为0.5时加入1.0 mmol/L IPTG(Sangon),继续诱导培养6 h,离心收集菌体,-20 ℃ 保存。菌体用2.0 mL结合缓冲液(20 mmol/L sodium phosphate, 500 mmol/L NaCl, 20 mmol/L imidazole, pH7.4)(Sangon)悬浮,在冰浴上超声波处理,收集裂解液并置 -80 ℃保存。上清液用结合缓冲液稀释后,再用Ni柱(GE Healthcare)于4 ℃ 中进一步纯化。
1.4 蛋白质凝胶电泳
十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis,SDS-PAGE):弃培养基后,培养细胞用预冷PBS洗3次,加入500 μL 1×IPP细胞裂解液 〔50 mmol/L Tris-HCl(pH 8.0),150 mmol/L NaCl, 1% NP40(Roche),1×蛋白酶混合抑制剂(Roche)〕,放入T25/D60培养皿,冰上置5 min,4 ℃ 中 12 000 rpm离心5 min,收集上清液并分装(100 μL),-80 ℃ 保存。电泳前取细胞裂解产物加等体积的2×SDS-PAGE上样缓冲液,煮沸5~8 min。冷却后 12 000 rpm离心2 min。凝胶制备及电泳用Bio-Rad Mini装置完成,主要试剂购自Calbiochem和Amresco公司,分离胶浓度10%~12%,浓缩胶浓度5%。电泳条件:浓缩胶80 V 30 min,分离胶120 V 60 min,或全程恒流15 mA/gel。电泳后凝胶用去离子水洗3次,转移至PVDF膜(Bio-Rad)并用抗体检测(见下),或用考马斯亮蓝R250染色后直接拍照分析或做蛋白质打点质谱鉴定。凝胶图片用Adobe PhotoShop CS5软件调整参数,然后取目的蛋白点,最后用Image J软件进行灰度比较分析。
2D电泳:吸去培养液后,细胞用预冷PBS洗3次,加入1.0 mL 1×IPG裂解缓冲液〔7 mol/L urea(Sigma),2 mol/L thiourea(Sigma),4% CHAPS(Bio-Rad),0.5% bio-lyte 3-10 ampholytes(Bio-Rad),40 mmol/L DTT(Bio-Rad),1/100 (V/V) TURBO DNase I(Ambion)〕,放入T75/D100培养皿,冰上置15 min,4 ℃ 中 12 000 rpm离心5 min,收集上清液并分装(120 μL),-80 ℃ 保存。电泳前取120 μL IPG裂解样品,加入30 μL水化液(IPG 缓冲液 +0.002%溴酚蓝),室温平衡后吸取125 μL上清液并均匀加到7 cm胶条/槽(Bio-Rad)上,再用适量矿物油(Bio-Rad)完全覆盖胶条,移至等电聚焦仪Ettan IPGphorⅡ(Amersham Biosciences/GE Healthcare)上被动水化7 h。然后梯度电泳:50 V 7 h,100 V 7 h,300 V 30 min,1 000 V(G)30 min,5 000 V(G)90 min,5 000 V 60 min。电泳完成后取出胶条,用10 mL去离子水漂洗1次,加入5 mL平衡1液〔6 mol/L urea, 75 mmol/L Tri-HCl(pH 8.8),29.3%(V/V)glycerine,2% SDS,0.002%溴酚蓝,0.05 g DTT〕平衡5 min,用去离子水再漂洗1次,加入5 mL平衡2液〔6 mol/L urea, 75 mmol/L Tri-HCl(pH 8.8), 29.3%(V/V)glycerol, 2%SDS, 0.002%溴酚蓝, 0.125 g IAA(Bio-Rad)〕平衡5 min。将胶条放置前述SDS-PAGE装置分离胶上方(一侧放蛋白marker滤纸),加入琼脂糖封闭液(25 mmol/L Tris, 192 mmol/L glycine, 0.1% SDS, 0.5% agarose, 0.002%溴酚蓝),静置3~5 min后开始电泳。
1.5 免疫印迹及抗体
蛋白质转印用Bio-Rad膜电转移装置(Mini Trans-Blot®)完成。PVDF膜用甲醇湿润后,与滤纸和海绵垫浸泡于转膜缓冲液(48 mmol/L Tris-39 mmol/L glycine, 0.037% SDS,20%(V/V) Methanol)中15 min以上,将电泳分离的凝胶另用新鲜转膜缓冲液浸泡,然后制备成胶膜“三明治”,叠层放入转膜槽中并设置降温装备,500 mA恒流电泳2 h;转膜完毕后取出PVDF膜,用PBS漂洗3次,做好标记和切割;用5%脱脂奶粉/PBS室温封闭1 h,用PBST(0.05% Tween-20/PBS)冲洗后转移至反应槽或塑封袋内;将特异性抗体用5%脱脂奶粉/PBST稀释好,加入反应槽中,4 ℃ 轻摇过夜;第2天用PBST漂洗,并加入用5%脱脂奶粉/PBST稀释好的HPR标记二抗,室温摇动孵育1 h;用PBST漂洗后加ECL(GE),并立即用X光片或化学发光成像仪(Tanon®4600)曝光。
本研究抗MHV-A59 N蛋白单克隆抗体2E6为本实验室制备[16],广谱磷酸化抗体anti-pSTY(SPM101)、磷酸激酶PKCa及S657磷酸化抗体〔均为兔单克隆抗体(RabMab®)〕购自Abcam,预染分子量标准(PageRuler®)购自Thermofisher,HRP-交联GAPDH鼠单克隆抗体购自Proteintech。
1.6 蛋白质质谱分析
凝胶考马斯亮蓝染色出现蛋白点或条带后,立即将蛋白质点小心割下,捣碎后置于96孔微孔板中。先用60 μL脱色液脱色,然后用去离子水洗2次至胶粒透明,再用60 μL乙腈干燥2次。加入消化液(含0.01 μg/μL胰蛋白酶的20 mmol/L NH4HCO3溶液)置室温孵育20 min,然后转移到37 ℃ 孵育过夜。第2天用60 μL提取液(含0.1%三氟乙酸的50%乙腈溶液)提取2次,合并上清液,在N2保护下干燥。用0.8 μL基质溶液重溶后点靶板。MALDI分析用 5 800 MALDI TOF/TOF分析仪(AB SCIEX)。设定每个点在700~4 000 m/z质谱范围内采用正离子反射模式获取一级质谱,激光累积1 000次激发。质谱(mass spectroscopy, MS)数据用内标进行校准。母离子按如下标准选择:每点最多选择12个母离子,信噪比最低设置为25,排除人角蛋白酶降解峰、胰酶自降解峰及基质峰。串联质谱采用2 500次激光累积和100分辨率的质量窗口(半峰全宽度)。碰撞能量设置为2 kV。连续线性质谱(MS/MS)数据采用默认校准。得到的数据用软件MASCOT(V2.3)进行检索,参数设定:NCBI蛋白库,胰酶酶切,一个漏切位点,一级质谱的容差为200 ppm,二级质谱的容差为0.8D。
2 结果
2.1 鼠冠状病毒核衣壳蛋白存在电荷非均一性和磷酸化
鼠冠状病毒MHV-A59 N蛋白理论上相对分子质量为49.7 kD, 454个氨基酸残基中含有41个丝氨酸、22个苏氨酸和11个酪氨酸,NetPhos 2.0预测有24种丝氨酸、5种苏氨酸和1种酪氨酸共30个潜在的磷酸化位点,实际质谱测定位6个,其中S170/S171修饰是随机的[17]。结果发现病毒感染后(24~36 h)neuro-2a细胞表达的N蛋白并不是单一电泳迁移条带,而感染后早期(12 h)的N蛋白为单一条带(图1A)。免疫印迹试验显示,N蛋白主要形成相对分子量40~55 kD的3种条带,而大肠埃希菌表达的N蛋白及初步纯化的病毒颗粒中N蛋白与之不完全一致,即原核细胞中缺少相对分子量较小的条带,大部分情况下只有相对分子量最大的单一条带[16];而初步纯化的病毒颗粒中相对分子量较小的条带明显增加,中间条带则相对较少(图1B)。
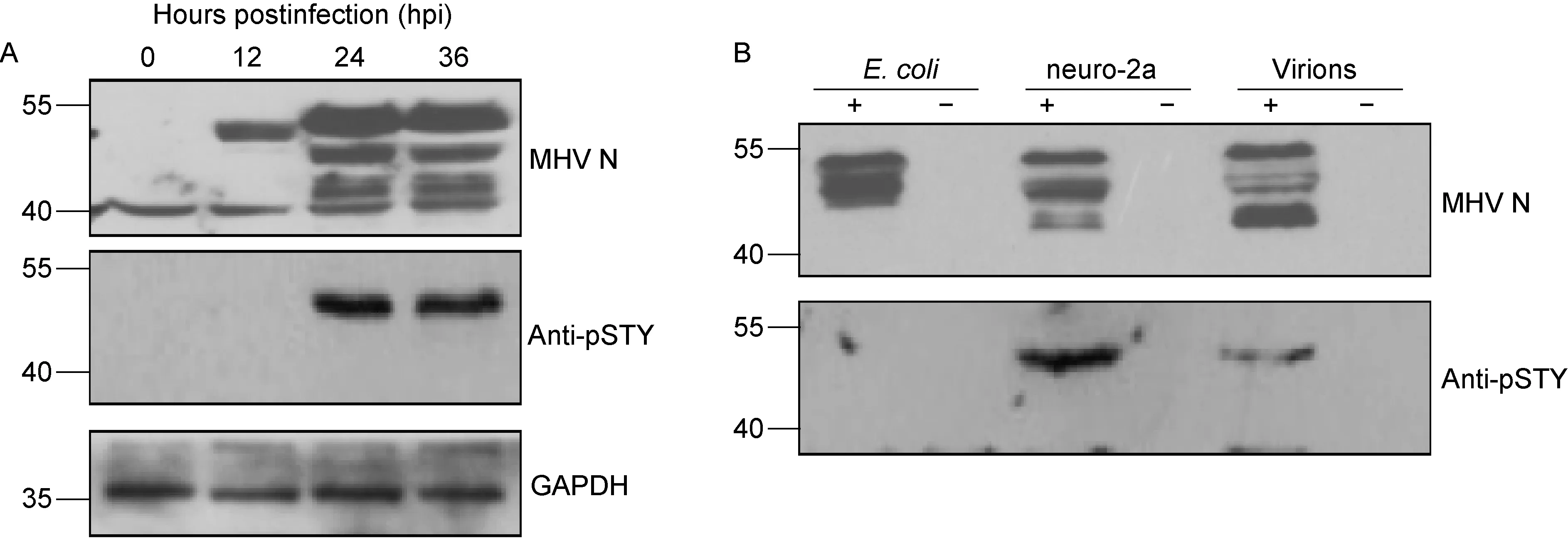
A:Time-course expression and phosphorylation of MHV-A59 N protein in neuro-2a cells. hpi: hours postinfection. B: N protein and phosphorylation of MHV-A59 from transformedEscherichiacolilysate, virus-infected neuro-2a lysate and PEG-purified virions.
图1 鼠冠状病毒核衣壳蛋白的非均一性及磷酸化
Fig.1 Heterogeneity and phosphorylation of murine coronavirus N protein
为了确定这种电泳迁移率的变化与蛋白磷酸化的关系,采用广谱抗磷酸化抗体(Anti-pSTY)检测了相应的条带。免疫印迹试验显示,感染早期病毒表达的单一条带即N蛋白不与广谱磷酸化抗体发生反应,原核载体pET-28a表达的N蛋白也不与广谱磷酸化抗体发生反应,只有来源于病毒感染后期及病毒颗粒中N蛋白能与广谱磷酸化抗体反应,但病毒颗粒中N蛋白反应明显弱于细胞表达的N蛋白(图1B)。结果提示磷酸化可能与N蛋白的降解有关,细胞中和病毒颗粒中N蛋白的磷酸化程度也不一样,虽然在病毒感染鼠17Cl1细胞时两者的磷酸化位点是相同的[17]。
2.2 鼠冠状病毒复制及N蛋白电负性受抑制剂2-BP和MG-132的影响
为了进一步分析MHV-A59 N蛋白非均一性与其磷酸化及其他修饰关联,我们选用了2种底物模拟竞争性小分子化合物2-BP和MG-132,对病毒复制及其N蛋白合成进行干预,前者抑制蛋白脂酰化修饰和膜结合功能,后者抑制蛋白降解作用[18-19]。用1.0 MOI MHV-A59感染neuro-2a细胞1 h,然后分别用含25 μmol/L 2-BP和10 μmol/L MG-132的低血清培养基(2% FBS/VP-SFM)替换,收集感染后24 h的培养上清液和细胞IPG裂解物,再用噬斑法测定活病毒滴度,2D电泳分离N蛋白并对抗体2E6进行检测。结果显示,2-BP抑制鼠冠状病毒复制明显强于MG-132(图2A);而鼠冠状病毒N蛋白并未集中在某一等电点上,而是呈现连续性分布,但相对分子质量变化不明显(图2B)。令人意外的是,2-BP处理导致了N蛋白集中在低等电点一侧,而MG-132处理则使更多N蛋白迁移到高等电点一侧(图2B)。这一结果至少说明,2-BP处理细胞使病毒N蛋白表面携带更少的正电荷,而MG-132处理则使N蛋白表面携带更多的正电荷,与其磷酸化修饰后果表现一致。同时,更多磷酸化的N蛋白有利于鼠冠状病毒的复制而提高了其滴度。
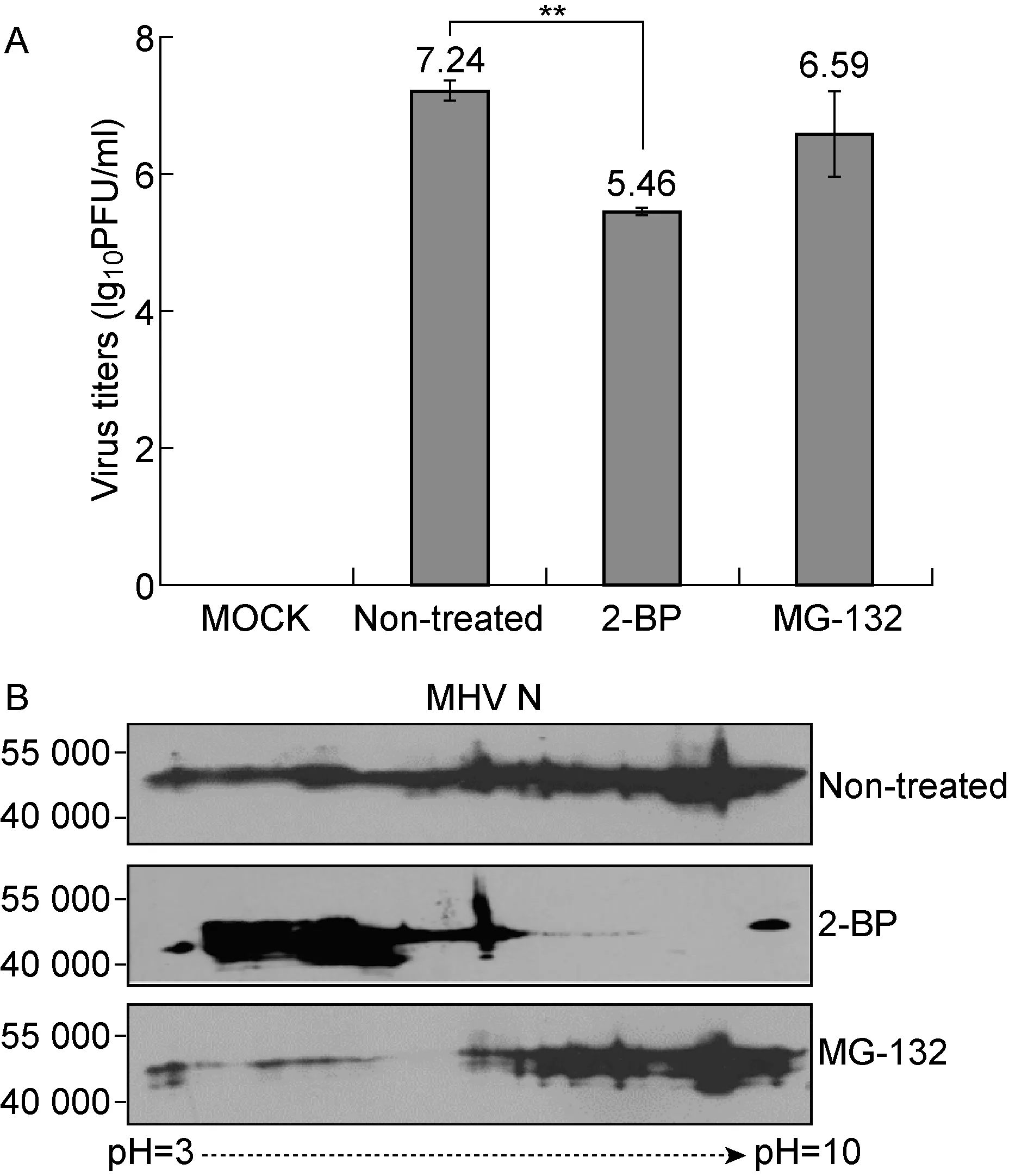
A:Effects of 2-BP and MG-132 on MHV-A59 titers (log10PFU/ml) in the supernatants of Neuro-2a cell culture at 24 hpi. **:P<0.01. B:Electromobility and reactivity to monoclonal antibody 2E6 of MHV-A59 N protein undergone 2D electrophoresis. Three panels are corresponding to the bottom three panels in Fig.4.
图2 2-BP和MG-132对鼠冠状病毒复制及核衣壳蛋白电负性的作用
Fig.2 Effects of 2-BP and MG-132 on the replication and N protein electronegativity of murine coronavirus
2.3 PKCa-S657磷酸化水平影响鼠冠状病毒的复制及N蛋白磷酸化
为了确定2-BP和MG-132处理对鼠冠状病毒MHV-A59 N蛋白电负性的影响与磷酸化的关系,首先测定了小分子抑制剂处理的感染细胞中N蛋白的磷酸化程度,然后分析两者与细胞中优势PKCα的相关性。实验结果显示,2-BP处理的病毒感染细胞中N蛋白的合成减少,同时其磷酸化明显降低(图3A);MG-132处理的病毒感染细胞中N蛋白合成和磷酸化也降低,但降低程度弱于2-BP处理组(图3A),两者的下降趋势与上清液中病毒颗粒滴度下降程度基本一致(图2A)。另外,病毒感染和两种抑制剂对未激活的PKCα无影响,但对其疏水基序磷酸化位点S657活化的PKCα(PKCα-pS657)则有不同程度的调节作用:病毒感染导致了PKCα-p S657明显下降,2-BP也抑制PKCα-pS657,但两者叠加却作用不明显(图3B);而MG-132的作用则不同, 虽然对细胞本身PKCα-pS657的作用无明显提高,但能明显拮抗病毒复制对PKCα-pS657的抑制作用(图3B)。
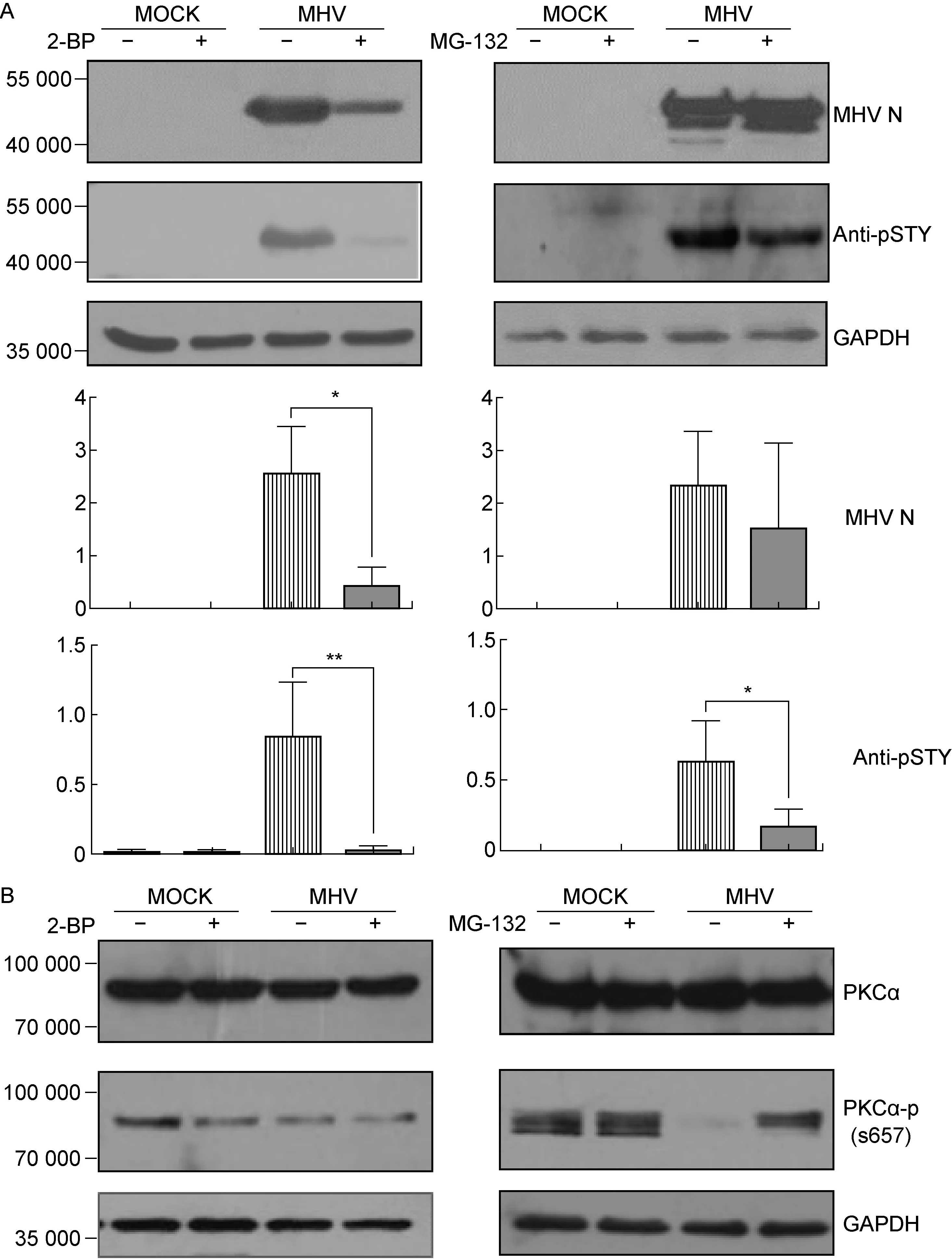
A:Effects of 2-BP and MG-132 on expression and phosphorylation of MHV N protein in neuro-2a cells at 24 hpi with MHV-A59. The histograms represent the mean distribution and test of the gray values from triple experiments.*:P<0.05;**:P<0.01. B:Effects of 2-BP and MG-132 on PKCα phosphorylation in infected neuro-2a cells at 24 hpi with MHV-A59.
图3 2-BP和MG-132对鼠冠状病毒核衣壳蛋白磷酸化的影响
Fig.3 Effects of 2-BP and MG-132 on the phosphorylation of murine coronavirus N protein
2.4 ERAD相关蛋白稳定有利于鼠冠状病毒的复制
图2和图3结果显示,鼠冠状病毒复制导致细胞中磷酸化PKCα的消耗,MG-132拮抗这种消耗,从而保持了一定的病毒复制水平;而2-BP因抑制了细胞的PKCα磷酸化水平而病毒复制明显降低。为了获得参与调节鼠冠状病毒N蛋白及其磷酸化的细胞蛋白相关线索,对病毒感染及2种小分子抑制剂处理的neuro-2a细胞进行了2D分离及对部分优势蛋白的质谱鉴定(图4)。结果显示,其一,病毒感染后合成的大量N蛋白沿约50 kD迁延分布,与特异性单克隆抗体2E6反应强烈(图2B);其二,病毒感染后一部分细胞蛋白合成被抑制,同时2-BP和MG-132也能抑制部分蛋白合成,但两者形成的蛋白谱存在较明显差异(图4)。
如图4所示,2-BP和MG-132两者对细胞的结构蛋白β-actin(黑圈)和α-enolase(红箭头)只有轻度的抑制。鼠冠状病毒MHV-A59感染后α-enolase明显减少,但对β-actin水平影响并不明显,因此β-actin可以作为其他细胞蛋白变化的参考标准。我们观察到与蛋白折叠和降解相关的辅助蛋白如HSP70-protein 9(绿箭头)和Calrecticulin(蓝箭头)等的变化与抑制剂密切相关。该结果提示2-BP不但对细胞蛋白S-棕榈酰化有抑制,而且间接抑制内质网转运功能相关蛋白;MG-132则有利于维持这两种蛋白的稳定。
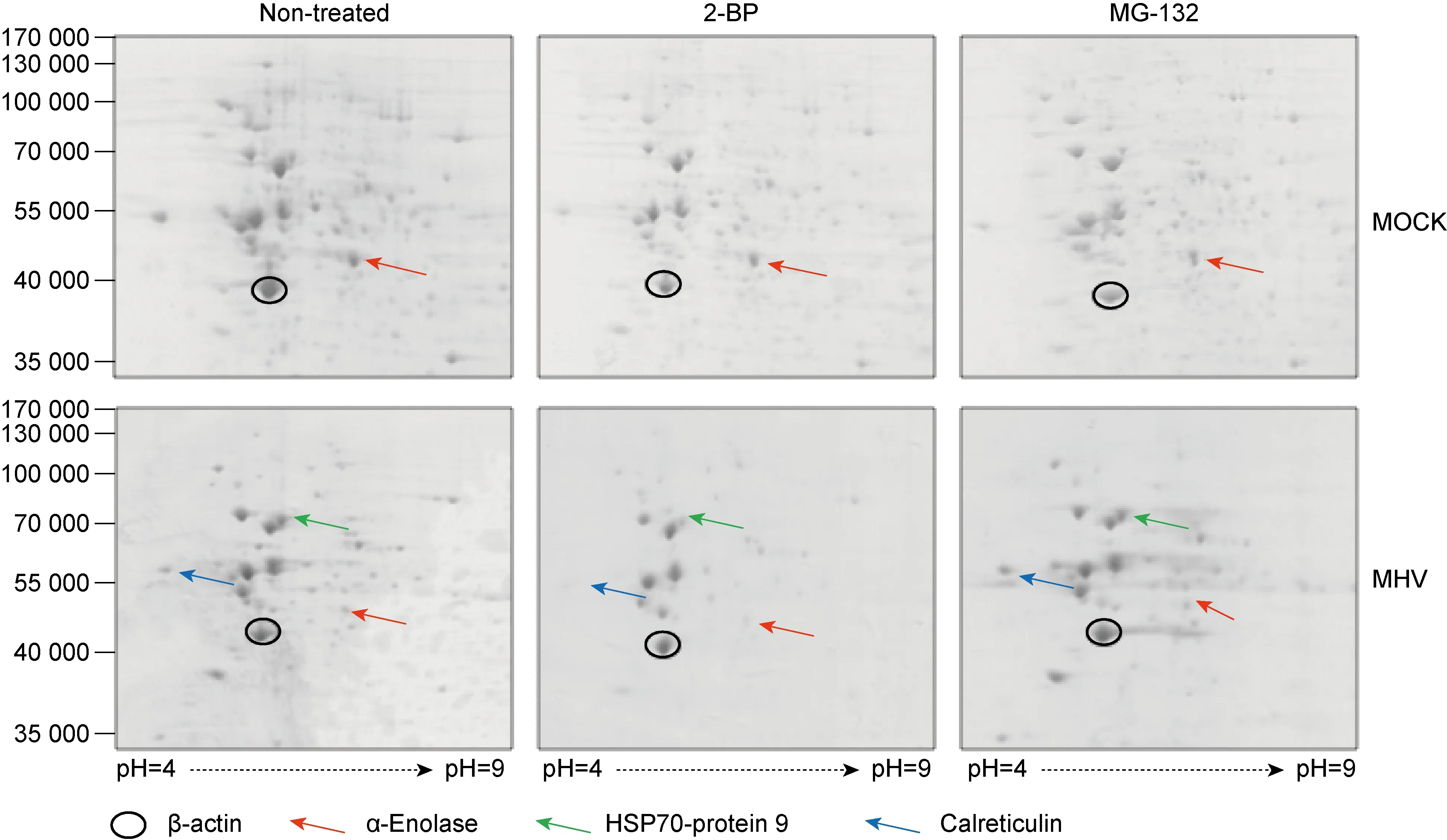
图4 2-BP和MG-132对鼠冠状病毒感染neuro-2a细胞优势蛋白表达的影响
Fig.4 Effects of 2-BP and MG-132 on the expression of dominant proteins of neuro-2a cells infected with murine coronavirus
3 讨论
neuro-2a细胞是一种特化的鼠神经母细胞瘤细胞系,产生大量微管蛋白,早期用于神经纤维蛋白的合成、装配和周转的研究,证明了细胞朊蛋白的聚集作用[14]。我们前期的工作发现该细胞系表面能稳定表达鼠冠状病毒受体分子mCEACAM1a, 支持MHV-A59的复制,并产生多种信号转导途径的分子应答[20]。另外,该细胞易于转染,能有效表达外源基因,比较适合鼠冠状病毒各种蛋白结构与功能的分析[21]。值得注意的是,胎牛血清白蛋白对稳定细胞生长相关因子非常重要,其本身也是促进细胞生长的重要成分;但与小分子2-BP或MG-132结合会影响其作用,因此不能用含高浓度血清的完全培养基分析其作用。neuro-2a细胞在无胎牛血清的DMEM中生长明显减慢,且形态大小不均。为此我们选用商品化无血清培养基(VP-SFM)添加约2%胎牛血清来替代,10 μmol/L MG-132和25 μmol/L 2-BP均获得很好的作用效果(图2~4)。
鼠冠状病毒N蛋白形成NTD和CTD两个高度保守结构域,前者主要结合调控病毒RNA复制,后者与自身同源聚集相关[22]。鼠冠状病毒N蛋白形成特征性的磷酸化位点2个热点簇,一个位于NTD与CTD之间的SR富含区,包括S162/S170/T177;另一个位于CTD与C端N3功能域之间,包括S389/S424/T428[17]。Wu等证明GSK3催化SARS-CoV N蛋白S177和MHV-JHM N蛋白S197磷酸化有利于病毒长模板RNA转录[8-9]。实验中我们注意到,低等电点一侧的N蛋白主要包含大相对分子质量的未磷酸化组分,与病毒复制早期合成N蛋白一致;而高等电点一侧的大相对分子质量条带除了含有磷酸化和未磷酸化者外,还包含了中间或小相对分子质量未磷酸化组分,与病毒复制的后期合成一致(图 1和图 2B)。另外,广谱磷酸化抗体Anti-pS/T/Y只与相对分子质量最大的N蛋白条带反应,单克隆抗体2E6能与3种不同相对分子质量的N蛋白条带反应(图1和图2B)。前者一般不能识别磷酸化位点上下游序列,后者识别区位于MHV-A59 N蛋白的NTD与CTD之间[16]。这些结果说明,相对分子质量中等和较小的N蛋白条带有可能是C端磷酸化位点之后序列被降解而形成,其中包含了具有与病毒其他结构蛋白相互作用功能的N3结构域[22],说明装配至成熟病毒颗粒N蛋白可能存在多种修饰形态。但是仅通过电泳结果和蛋白棕酰化抑制剂2-BP和蛋白酶体抑制剂MG-132处理不能推断N蛋白电荷不均一性由其磷酸化引起。为了获得电荷负载变化与磷酸化的直接关联数据,我们试图用抗体2E6通过免疫沉淀浓集3种不同处理的病毒N蛋白,再用HPLC-ESI-MS/MS对磷酸化位点进行定位[17],但因所得蛋白的量和纯度有限及MS技术问题未获成功,此实验仍在改进之中。另外,目前PKC抑制剂无型别特异性,对N蛋白磷酸化及电负性影响特异性大部分不如2-BP和MG-132。病毒N蛋白磷酸化机制和效应目前仍未完全清楚,研究技术手段相对缺乏,本文提示电负性作为磷酸化修饰潜在指标值得进一步探索,虽然两者之间相关性仍需更可靠的证据。
PKCα磷酸化是PKCα激活的基本条件之一,其C端结构域中有3个位点可依次被磷酸化,首先位于激活环(activation loop)内的T497能被PDK1磷酸化,随后转折基序(turn motif)上的T638和疏水基序(hydrophoblic motif)上的S657发生自动磷酸化[10]。MG-132在较高浓度时,也具有抑制细胞磷酸化的作用[23]。因此,PKCα-p S657代表了活化的PKCα。据此我们认为:①鼠冠状病毒复制及其N蛋白的磷酸化依赖于活化状态的PKCα;②2-BP抑制细胞活化状态的PKCα-pS657产生而导致的病毒复制及其N蛋白的合成和磷酸化降低可能是通过抑制细胞蛋白脂酰化所致,因为膜定位能力对PKCα激活非常关键;③MG-132可能通过抑制磷酸化N蛋白的降解维持病毒的复制和装配。
MG-132为醛基化亮氨酸三肽类似物(Z-Leu-Leu-leucinal),是一种特异的、可逆的和渗透性强的20S蛋白酶体抑制剂,其导致不同种类肿瘤细胞凋亡与剂量有关[19]。我们发现10 μmol/L MG-132对鼠neuro-2a细胞不产生明显凋亡作用,但能有效延缓鼠冠状病毒感染导致的磷酸化PKCα及其他优势蛋白质的降低,处理后病毒复制仍可维持在较高的水平(图 3、图 4)。这一结果与曾报道的MG-132抑制病毒复制和致病性并不完全一致,可能与抑制剂浓度和细胞系不同有关[24-25]。但结合2-BP同时对鼠冠状病毒复制及细胞ERAD相关蛋白组分抑制现象推测:MG-132保护了ER结合蛋白不被蛋白酶体破坏,有利于复制后期病毒颗粒装配所必需的双层膜囊泡的形成[26]。ER蛋白是否参与病毒膜相关蛋白S、M、E合成与修饰以及细胞PKCα的活化过程,再通过N蛋白磷酸化调节病毒复制,仍待进一步实验探索。
2-BP及其衍生物由于能特异性结合含DHHC基序的蛋白脂酰转移酶(protein acyl transferases,PATs)而广泛用于蛋白棕榈酰化的研究[18]。病毒Ⅰ型膜融合蛋白易发生棕榈酰化,因而2-BP能干扰这些病毒的复制,包括流感病毒HA、人类免疫缺陷病毒gp41、冠状病毒S蛋白等[27]。但2-BP也能非特异性且不可逆地抑制一些细胞内膜结合蛋白如Src家族成员的磷酸化而影响细胞代谢过程[28]。我们发现培养基中含25 μmol/L 2-BP能够部分抑制细胞膜蛋白和鼠冠状病毒N蛋白合成及PKCα磷酸化(图 2~4)。2-BP在10~50 μmol/L对病毒的抑制作用并无明显剂量依赖性,但低于10 μmol/L作用不明显,而高于50 μmol/L会导致neuro-2a细胞形态改变并凋亡(未发表数据)。因此,我们推测2-BP导致的病毒复制不仅直接作用于病毒S蛋白的棕榈酰化,也可能因细胞HSP70和HSP90/cdc37等伴侣分子减少间接抑制了PKCα磷酸化[29],后者导致磷酸化N蛋白参与的复制酶-转录酶复合物(replicase-transcriptase complex, RTC)的结构形成、RNA合成等功能受损[29-30]。另外,冠状病毒RTC也可能具有活化的PKC受体(receptors for activated PKC,RACK)类似功能,支持PKCα磷酸化,但复制后期大量包膜蛋白尤其是糖基化S蛋白的累积导致伴侣蛋白HSP70等消耗,反馈抑制PKC-N磷酸化,启动病毒进入颗粒装配阶段。
猜你喜欢
计算机应用与软件(2022年6期)2022-07-12
油气地质与采收率(2022年3期)2022-05-20
自然灾害学报(2022年2期)2022-05-10
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
天津医科大学学报(2021年4期)2021-08-21
中国学校体育(2021年10期)2021-04-26
现代临床医学(2021年2期)2021-03-29
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
