
非天然氨基酸定点偶联抗人类表皮生长因子受体2-抗体偶联药物的药理学活性
2019-10-28 09:40梁学军宫丽颖周德敏祝静静
北京大学学报(医学版) 2019年5期
梁学军,宫丽颖,周 菲,周德敏,祝静静△
(1. 浙江新码生物医药有限公司,浙江绍兴 312000; 2. 北京大学药学院化学生物学系,北京 100871)
抗体偶联药物(antibody drug conjugate, ADC)是利用单克隆抗体实现细胞毒性药物靶向传递到抗原表达细胞的一类药物[1]。ADC具有单克隆抗体(monoclonal antibody, mAb)的靶向特异性,同时它又通过偶联放射性核素或细胞毒药物进而增强治疗活性,在恶性肿瘤的治疗过程中只需要单一给药即可达到理想的治疗效果。ADC一般由单克隆抗体、细胞毒性分子和连接子三部分组成。与单抗药物不同,ADC药物的研发除了选择可以识别细胞表面抗原并被细胞内吞的单克隆抗体,还需要综合考虑选用的细胞毒性分子及偶联方法[1-2]。传统的偶联方法主要是依靠天然氨基酸中的赖氨酸或半胱氨酸侧链上活泼的氨基和巯基,对蛋白药物进行偶联[3-4],但是这些传统连接方法的缺点是偶联位点及偶联的小分子数目缺少特异性。目前正在研究的ADC药物,多数是通过半胱氨酸或赖氨酸对单克隆抗体和毒素进行偶联,得到的ADC是非均一的混合物,例如曲妥珠单抗-美坦新偶联物(ado-trastuzumab emtansine)中含有偶联0~8个毒性分子的ADC,平均3.5个毒性分子左右[4-5];本妥昔单抗中含有偶联0~8个毒性分子的ADC,平均4个毒性分子左右[5],因此传统的偶联方法很难控制药物的均一性,且不同有效载荷的ADC的药效、生物安全性、动力学参数等也各不相同[3],而目前ADC的定点偶联技术可以有效地解决这类问题[6]。
实现定点偶联的方法有多种,例如半胱氨酸突变、酶催化连接、糖工程、引入非天然氨基酸等[7-11]。非天然氨基酸技术实现蛋白质的定点修饰技术最早由Schultz实验室开发,Wang等[12]在细胞或细菌内引入能够特异性识别非天然氨基酸的外源tRNA以及与之相对应的氨酰tRNA合成酶,该酶能够催化特定非天然氨基酸与该tRNA结合,然后进入核糖体,并通过识别mRNA上的琥珀终止密码子UAG,将非天然氨基酸引入到增长肽链的相对应位置,进而实现定点偶联。
人类表皮生长因子受体2(human epidermal growth factor receptor 2, HER2)属于表皮生长因子受体家族成员,具有酪氨酸激酶活性。HER2过度表达与侵袭性肿瘤的发生及发展具有紧密的关系。研究表明,在20%~30%的乳腺癌患者中有HER2高表达[13],同时在胃癌、卵巢癌、子宫癌患者中也发现有HER2高表达[14]。开发针对HER2高表达的药物,对于多种恶性肿瘤的治疗具有重要的意义,本研究使用的抗HER2-ADC为通过非天然氨基酸定点偶联技术获得的均一性良好的抗体偶联药物。该抗HER2-ADC中的单克隆抗体蛋白序列与临床上用于治疗HER2阳性肿瘤的曲妥珠单抗(trastuzu-mab)序列类似,通过每个抗HER2单克隆抗体分子中引入的非天然氨基酸pAF与毒性分子AS269的特异反应,得到定点定量偶联的抗HER2-ADC。本研究旨在研究抗HER2-ADC对HER2不同表达量的肿瘤细胞的增殖抑制活性及肿瘤异体移植模型的抗肿瘤活性,探讨其药理活性,为其在乳腺癌、胃癌和卵巢癌的临床研究提供数据支持。
1 资料与方法
1.1 细胞系和小鼠
人乳腺癌细胞系BT-474、HCC1954、MCF-7、MDA-MB-231、MDA-MB-468、SK-BR-3,人肺癌细胞系Calu-3,人胃癌细胞系NCI-N87,人卵巢癌细胞系SK-OV-3购自美国ATCC公司。实验小鼠购自上海西普尔-必凯实验动物有限公司。
1.2 试剂、材料和仪器设备
无水枸橼酸购自美国Sigma-Aldrich公司,QIFI试剂盒购自美国Agilent Technologies公司,CellTiter-Glo购自美国Promega公司,Pacific OrangeTMProtein Labeling Kit和DRAQ5购自美国Thermo Fisher Scientific 公司,二水合枸橼酸三钠、组氨酸、盐酸组氨酸一水合物和聚山梨酯80购自美国J.T.Baker公司,海藻糖二水合物购自日本Hayashibara公司,曲妥珠单抗和曲妥珠单抗-美坦新偶联物购自瑞士Roche公司,紫杉醇(paclitaxel)注射液购自扬子江药业集团,17β-雌二醇片购自美国Innovative Research公司,抗HER2-ADC、AS269和pAF-AS269由浙江新码生物医药有限公司委托药明康德新药开发有限公司生产,疏水色谱柱购自日本Tosoh公司。流式细胞仪(FACSCanto Ⅱ)购自美国Becton Dickinson公司,荧光共聚焦显微镜(UltraVIEWVoX)购自美国Perkin Elmer公司。
1.3 偶联反应
将含有非天然氨基酸的抗HER2 mAb溶液和毒性分子AS269水溶液混合,加入枸橼酸缓冲液(包括无水枸橼酸和二水合枸橼酸三钠),调节pH至4.0,室温保温偶联48 h。偶联完成后,再加入等量的pH 6.0的组氨酸缓冲液(包括组氨酸和盐酸组氨酸一水合物)淬灭反应,然后用组氨酸溶液对淬灭后的反应液进行超滤换液,去除残留小分子。超滤后的溶液添加海藻糖和聚山梨酯80,得到ADC原液抗HER2-ADC。抗HER2-ADC的偶联率(drug to antibody ratio, DAR)通过高效疏水作用色谱法检测得到。实验采用直径 4.6 mm、长10 cm、孔径2.5 μm的疏水色谱柱,检测波长为280 nm。DAR的计算公式为:DAR=(1Drug%+2×2Drugs%)/(mAb%+1Drug%+2Drugs%),mAb%、1Drug%和2Drugs%分别为未偶联、偶联了1个和2个AS269的mAb峰面积占总峰面积的百分比。
1.4 流式细胞术检测HER2在9种肿瘤细胞中的表达
将BT-474、Calu-3、MCF-7、MDA-MB-231、MDA-MB-468、SK-BR-3、SK-OV-3、HCC1954、NCI-N87共9种肿瘤细胞按照各自需要的培养条件在37 ℃、5%(体积分数) CO2的培养箱中进行培养[15-19],细胞铺满培养瓶的70%至80%时用于试验。收集细胞之后,根据QIFI试剂盒的说明,对细胞进行处理,然后通过流式细胞仪进行检测,根据试剂盒提供的数据分析方法使用流式细胞数据分析软件FlowJo来计算各细胞系HER2受体的数量。
1.5 5种药物对9种肿瘤细胞的增殖抑制活性
实验采用9种不同HER2表达的肿瘤细胞(包括源自乳腺癌、肺癌、卵巢癌和胃癌的肿瘤细胞),考察抗HER2-ADC、AS269、pAF-AS269、曲妥珠单抗-美坦新偶联物和紫杉醇对细胞的增殖抑制活性。AS269为含有四个乙二醇片段的毒性小分子,是海兔毒素系列衍生物[20]。pAF-AS269为带着非天然氨基酸pAF的AS269,是抗HER2-ADC在体内代谢后的产物。9种肿瘤细胞按照第1.4小节同样的方法进行培养,铺满培养瓶的70%至80%时用于试验。分别将细胞密度调整到MCF-7和MDA-MB-231为2.22×104个/mL,SK-OV-3为3.33×104个/mL,NCI-N87和Calu-3为4.44×104个/mL,HCC1954为5.56×104个/mL,MDA-MB-468、SK-BR-3和BT-474为6.66×104个/mL。对抗HER2-ADC、曲妥珠单抗-美坦新偶联物、AS269、pAF-AS269、紫杉醇5种药物进行梯度稀释,稀释后药物浓度范围是0.005~300 nmol/L。在96孔细胞培养板中分为3组:5种药物的试验组、溶媒对照组和空白对照组。试验组和溶媒对照组每孔分别加入90 μL的9种肿瘤细胞的细胞悬液,空白对照组中每孔加入90 μL不含细胞的培养液,将细胞培养板在37℃、5%CO2及100%相对湿度的培养箱中培养过夜。试验组分别加入10 μL稀释后的5种药物,溶媒对照和空白对照组每孔加入10 μL不含药物的细胞培养液。BT-474、MCF-7、HCC1954、MDA-MB-468和SK-BR-3这5种细胞继续培养72 h;Calu-3、MDA-MB-231、NCI-N87和SK-OV-3这4种细胞继续培养96 h,然后每孔加入100 μL CellTiter-Glo,检测发光信号,并计算待测药物的抑制率:抑制率=(相对光信号药物- 相对光信号空白对照)/(相对光信号溶媒对照-相对光信号空白对照)×100%。用GraphPad Prism5软件得到抑制曲线图并计算半数抑制浓度(half maximal inhibitory concentration,IC50)。
1.6 抗HER2-ADC的细胞内吞机制
使用Pacific OrangeTMProtein Labeling Kit试剂标记抗HER2-ADC并且纯化,获得荧光标记的抗HER2-ADC。HCC1954细胞按照第1.4小节同样的方法进行培养,待长到合适密度后,在96孔细胞培养板中每孔加入200 μL细胞悬液,细胞浓度为1×105个/mL,将细胞培养板在37℃、5%CO2及100%相对湿度的培养箱中培养过夜。用DRAQ5试剂对细胞核进行染色,室温放置15 min,然后加入5 mg/L Pacific OrangeTM试剂标记的抗HER2-ADC,用荧光共聚焦显微镜进行实时拍摄,拍摄过程维持细胞温度为37 ℃,按照DRAQ5与Pacific OrangeTM试剂要求,设置荧光共聚焦显微镜的激发光/发射光波长分别为640/681 nm和405/551 nm,总共记录时间为3.5 h。
1.7 小鼠模型体内药效学分析
以BT-474、HCC1954、MDA-MB-468、NCI-N87、SK-OV-3这5种肿瘤细胞异种移植的小鼠作为研究对象,分别考察抗HER2-ADC、曲妥珠单抗、曲妥珠单抗-美坦新偶联物和紫杉醇4种药物的体内抗癌效果。实验采用5~6周龄的BALB/c裸小鼠,实验动物的使用及福利遵照国际实验动物评估和认可委员会的规定执行。小鼠适应培养后,将5种肿瘤细胞分别皮下接种于每只小鼠的右后背。MDA-MB-468、SK-OV-3和BT-474细胞为0.1 mL的1×108个/mL细胞悬液与0.1 mL基质胶混合接种,其中BT-474细胞于接种前3天时,每只小鼠皮下接种1片17β-雌二醇(0.72 mg,60 d缓释)以供应肿瘤生长必需的雌激素;HCC1954细胞为0.1 mL的 5×107个/mL细胞悬液与0.1 mL基质胶混合接种;NCI-N87细胞为0.1 mL 的1×108个/mL细胞悬液直接接种。MDA-MB-468、SK-OV-3和BT-474肿瘤平均体积达到150~200 mm3时开始分别给药,每种药物10只小鼠;HCC1954肿瘤平均体积达到50~150 mm3时开始分别给药,每种药物10只小鼠;NCI-N87肿瘤平均体积达到150~200 mm3时开始分别给药,每种药物9只小鼠。抗HER2-ADC、曲妥珠单抗、曲妥珠单抗-美坦新偶联物和空白对照磷酸盐缓冲液经静脉注射给药,给药1次;紫杉醇经腹腔给药,每周两次,给药两周。
每天监测动物的健康状况及死亡情况,观察肿瘤生长和药物治疗对动物日常行为表现的影响:如行为活动、摄食摄水量(仅目测)、体质量变化(每周测量两次体质量)、外观体征或其它不正常情况。基于各组动物数量记录组内动物死亡数和副作用,每周两次用游标卡尺测量肿瘤直径,肿瘤体积的计算公式为:V=0.5a×b2(a和b分别表示肿瘤的长径和短径)。
根据肿瘤测量的结果计算出相对肿瘤体积(relative tumor volume,RTV),计算公式为RTV=Vt/V0,其中V0是分组给药时测量所得平均肿瘤体积,Vt为某一次测量时的平均肿瘤体积。化合物的抑瘤效果用相对肿瘤增殖率T/C(%)评价:T/C=TRTV/CRTV×100%(TRTV与CRTV分别为治疗组和空白对照组的相对肿瘤体积,TRTV与CRTV取同一天数据)。
1.8 统计学分析
每个组的每个时间点的肿瘤体积,结果用均数±标准误表示。应用Mann-WhitneyUtest法评估组间差异,用SPSS 17.0进行所有数据分析,P<0.05为差异有统计学意义。
2 结果
2.1 偶联获得抗HER2-ADC
本试验通过与引入两个非天然氨基酸pAF的抗HER2单克隆抗体中的特异性化学反应,在抗HER2单克隆抗体上偶联两个AS269(图1)。利用非天然氨基酸技术定点偶联获得的抗HER2-ADC,具有良好的均一性,其DAR理论值为2。在反应开始6 h后大部分抗HER2单克隆抗体已偶联了两个AS269;偶联48 h后通过高效疏水作用色谱法实际检测结果DAR为 1.95,其中2Drugs%为91.17%,1Drug%为4.69%,未偶联的mAb%为0.12%(图2)。
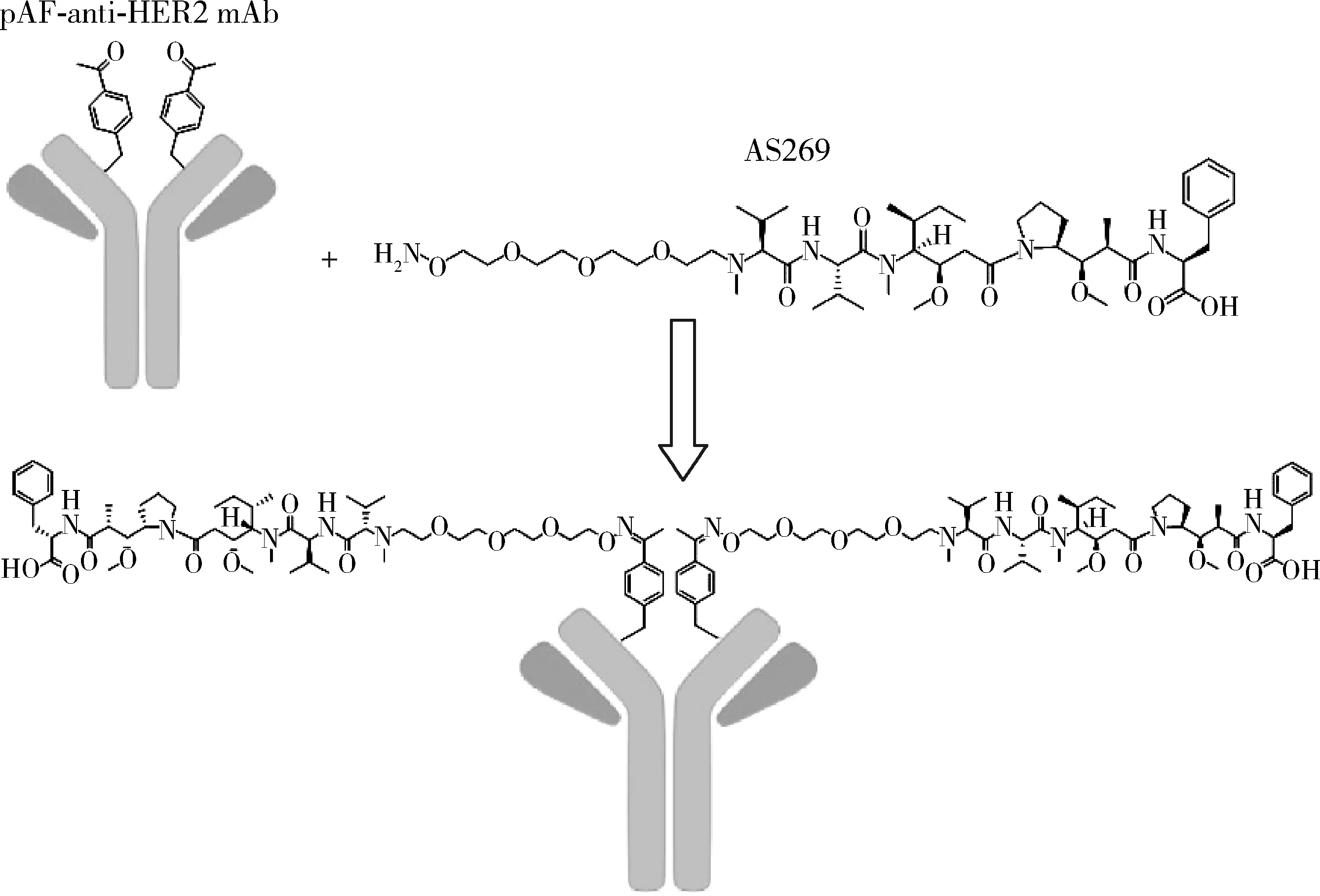
HER2, human epidermal growth factor receptor 2; mAb, monoclonal antibody.图1 抗HER2单克隆抗体和毒素AS269的定点偶联示意图Figure 1 Site specific conjugate anti-HER2 mAb and cytotoxic AS269

mAb, monoclonal antibody.图2 高效疏水作用色谱法检测抗HER2-ADC偶联率Figure 2 Conjugation rate profile of anti-HER2-ADC by high performance hydrophobic interaction chromatography
2.2 流式细胞术检测HER2在9种肿瘤细胞中的表达
根据QIFI试剂盒提供的方法计算每个细胞表面HER2受体密度,结果如表1所示:SK-BR-3、NCI-N87、SK-OV-3细胞系HER2表达量最高,BT-474、HCC1954、Calu-3细胞系也显示出较高的HER2表达,而MCF-7、MDA-MB-231细胞系HER2表达水平很低,在MDA-MB-468中几乎检测不到HER2受体的表达。
2.3 待测药物对9种肿瘤细胞增殖抑制活性分析
由表1所示,对于HER2受体高表达的乳腺癌细胞系,抗HER2-ADC的体外IC50分别为17 pmol/L(SK-BR-3)、50 pmol/L(BT-474)和125 pmol/L(HCC1954)。在6种HER2受体高表达的肿瘤细胞中,抗HER2-ADC均表现出比曲妥珠单抗-美坦新偶联物和紫杉醇更好的活性,尤其是在Calu-3细胞系中,抗HER2-ADC对细胞生长的抑制作用明显优于曲妥珠单抗-美坦新偶联物和紫杉醇。此外,所有细胞系对待测药物AS269与pAF-AS269均不敏感。由于AS269和pAF-AS269在中性条件下带负电荷,无法自由进出细胞,因此AS269与pAF-AS269对实验中的9种细胞均没有毒性。
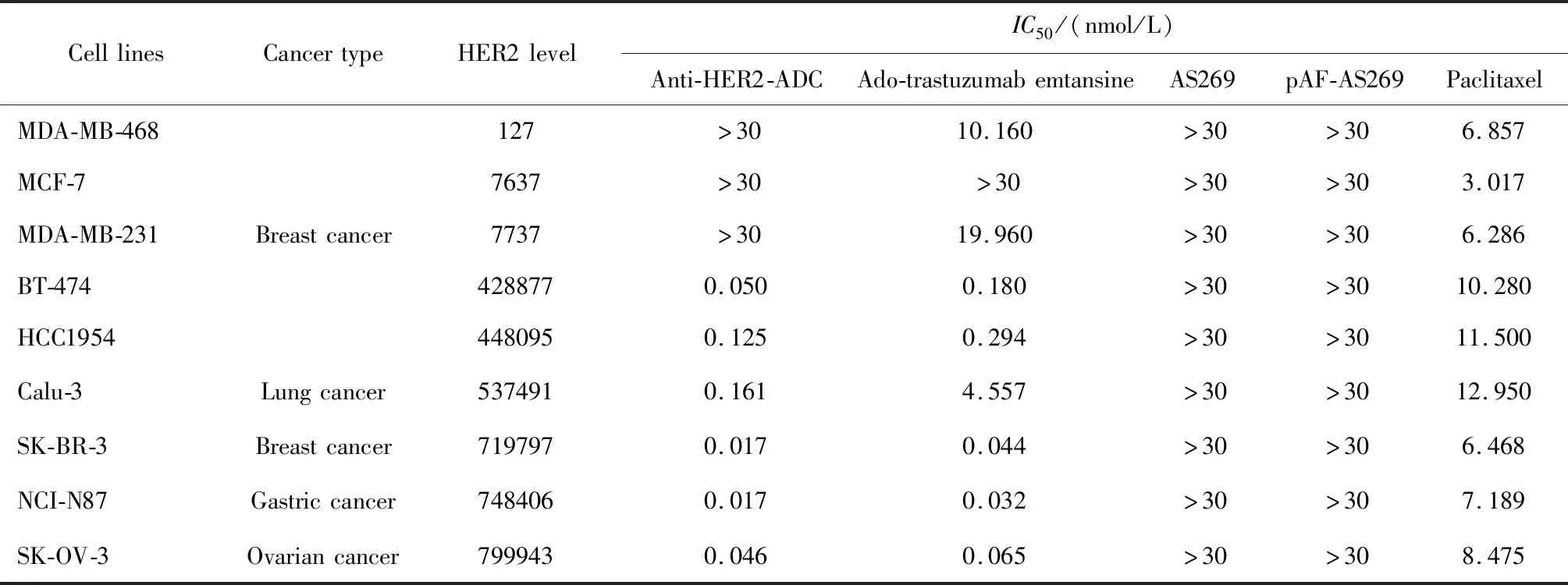
表1 细胞生长抑制活性Table 1 The inhibitory activities in cell proliferation assay
HER2, human epidermal growth factor receptor 2; mAb, monoclonal antibody; ADC, antibody drug conjugate.
2.4 抗HER2-ADC的细胞内吞机制
如图3所示,细胞核被DRAQ5染成紫色,加入Pacific OrangeTM标记的绿色抗HER2-ADC后0 h,未观测到培养液中游离的抗HER2-ADC。加入0.5 h后,相同视野可以观察到抗HER2-ADC在HCC1954细胞膜上富集,并有部分进入细胞内。随着时间的延长,抗HER2-ADC进入细胞内的量逐渐增加,在加入3.5 h后,细胞在药物作用下,细胞形态产生变化,开始萎缩,并有从培养皿脱落的现象。
2.5 小鼠模型体内药效学分析
图4在NCI-N87异种移植肿瘤模型中,对比了相同剂量的抗HER2-ADC、曲妥珠单抗和曲妥珠单抗-美坦新偶联物的抗肿瘤活性,结果显示抗HER2-ADC一次给药后,肿瘤体积在给药后4 d内略有增大,然后随着时间延长逐渐减小;曲妥珠单抗和曲妥珠单抗-美坦新偶联物在一次给药后,肿瘤体积在给药后18 d内增长缓慢,第18天后随着时间延长肿瘤体积迅速增加。在NCI-N87异种移植肿瘤模型中,与曲妥珠单抗及曲妥珠单抗-美坦新偶联物相比,相同剂量的抗HER2-ADC表现出更好的抗肿瘤活性,其T/C约为二者的1/30至1/20。
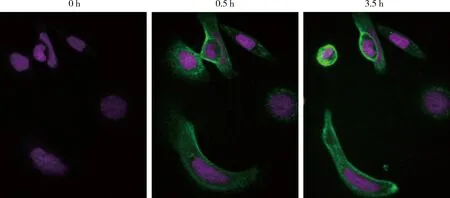
图3 DRAQ5染色和Pacific OrangeTM荧光标记后观察抗HER2-ADC在HCC1954细胞上的细胞内吞(×200)Figure 3 Internalization of anti-HER2-ADC in HCC1954 cell line after DRAQ5 staining and Pacific OrangeTM fluorescence labeling(×200)
表2汇总了不同浓度的抗HER2-ADC、曲妥珠单抗-美坦新偶联物、曲妥珠单抗和紫杉醇4种药物对5种异种移植肿瘤模型的动物体内药效,结果显示,对于HER2低表达细胞MDA-MB-468异种移植瘤模型,除了紫杉醇对其有抑制作用,其他靶向HER2的药物对其均未表现出显著抑制作用;而对于4种HER2高表达异种移植瘤模型,抗HER2-ADC与相同剂量的曲妥珠单抗-美坦新偶联物、曲妥珠单抗及更高剂量的紫杉醇相比,均表现出更佳的抗肿瘤活性。
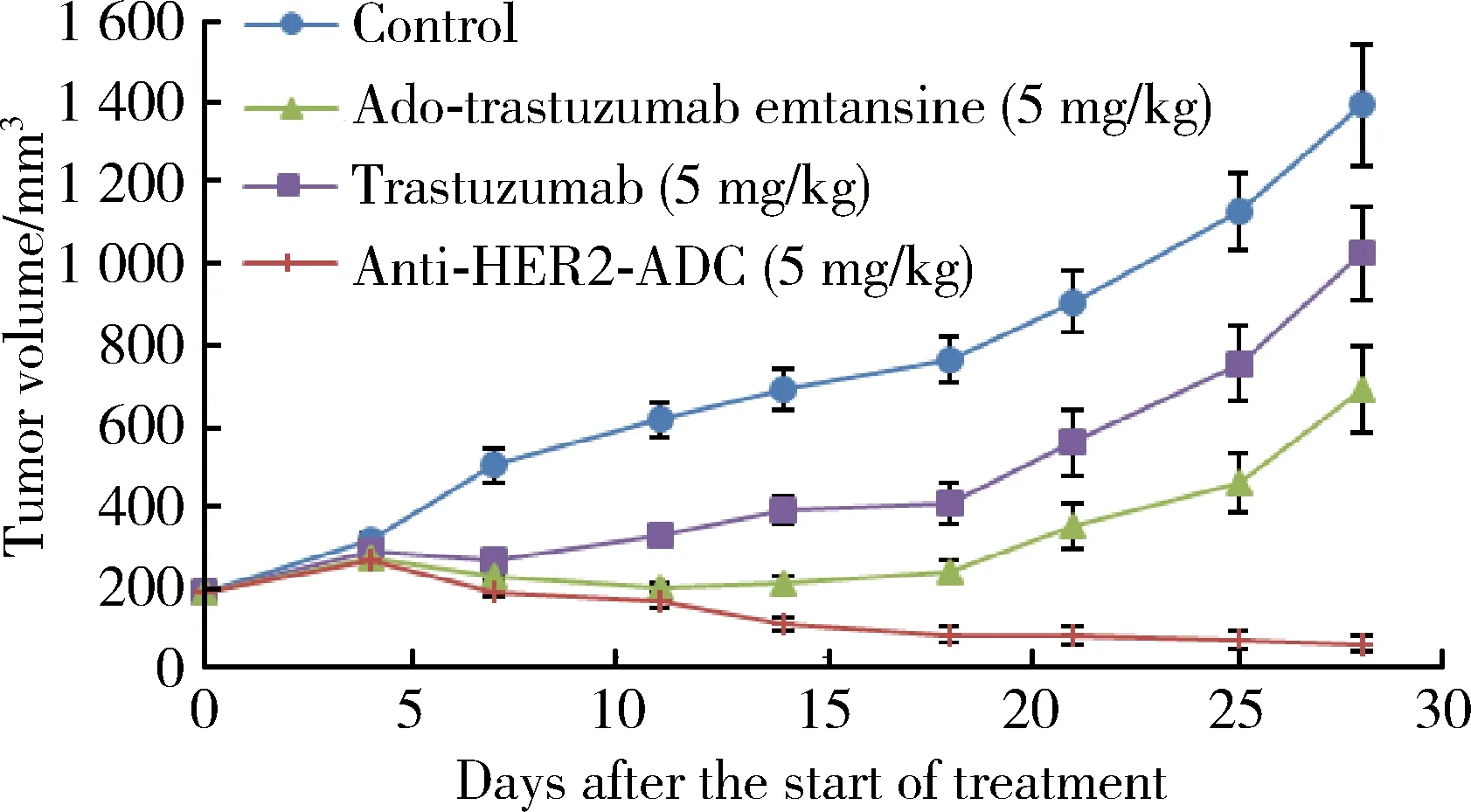
图4 NCI-N87异种移植瘤模型荷瘤鼠在给予药物后的肿瘤体积Figure 4 The tumor volume after administering drugs to xenograft model NCI-N87
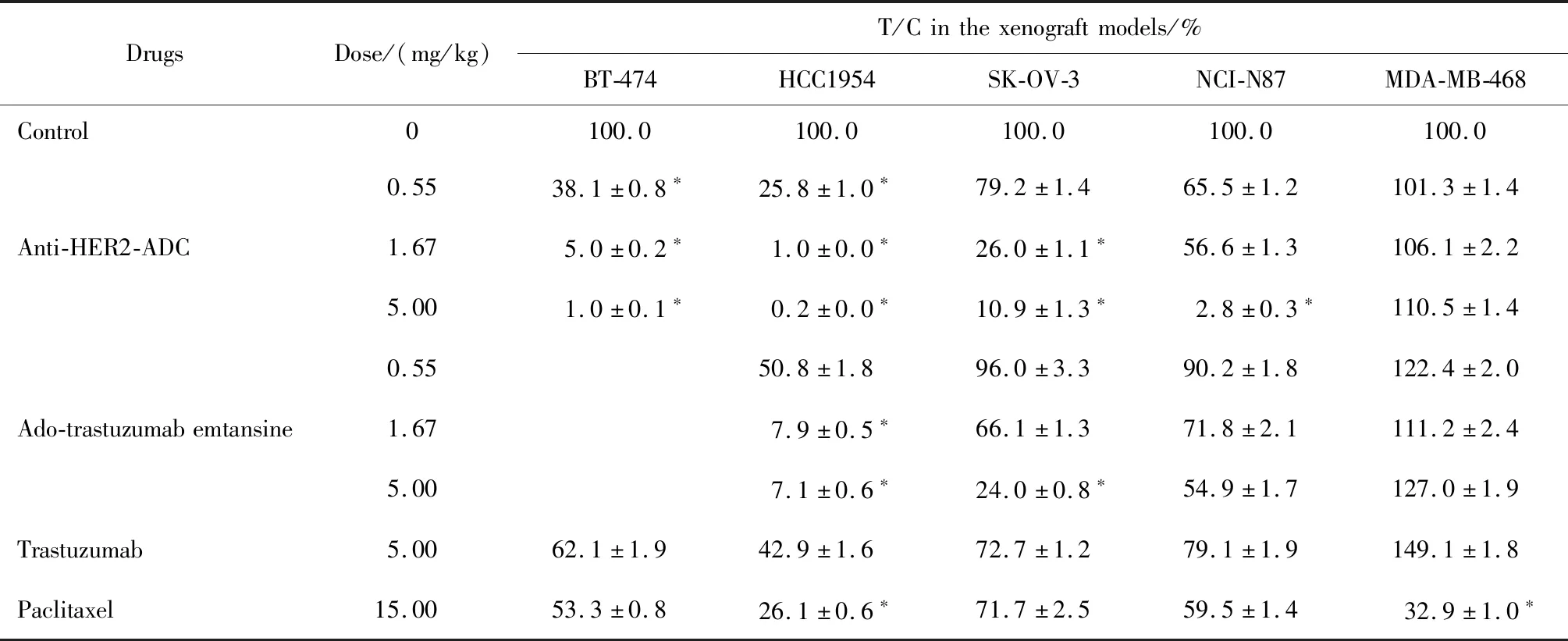
表2 小鼠异种移植肿瘤模型中待测药物的体内相对肿瘤体积增殖率Table 2 The relative tumor volume proliferation rate of test articles in the xenograft models
图5汇总了5种异种移植瘤模型给予不同剂量的HER2-ADC后的相对肿瘤增殖率,结果显示,在所有HER2高表达的异种移植瘤模型中,抗HER2-ADC表现出呈剂量依赖性的抗肿瘤活性,且注射剂量为5 mg/kg时,部分模型中表现出肿瘤的完全消退和治愈效果。此外,体质量作为毒性的间接检测指标,在所有移植瘤研究中进行定期监测。除了BT-474组有雌激素引起的体质量减轻或死亡,其他组均无异常报告,说明注射实验用的抗HER2-ADC剂量对免疫缺陷的肿瘤小鼠无明显毒性。

图5 异种移植瘤模型荷瘤鼠在给予抗HER2-ADC后的相对肿瘤体积增殖率Figure 5 The relative tumor volume proliferation rate after administering anti-HER2-ADC to xenograft models
3 讨论
毒素分子与抗体的偶联方法及连接子的选择是抗体偶联药物研究开发的两个难点。传统的偶联方法得到的ADC为非均一的混合物,各组分间药代动力学性质差异较大[21],而抗HER2-ADC采用非天然氨基酸技术实现定点偶联,获得的ADC均一性更好。另外,偶联使用的连接子分为可裂解和不可裂解两种[22],可裂解的连接子可以通过多种方法释放毒素分子,如酸性环境降解、溶酶体蛋白酶降解及谷胱甘肽降解等,增加了旁侧效应的可能性[23],而抗HER2-ADC使用不可裂解的连接子进行偶联,与前者相比,使用不可裂解的连接子偶联得到的ADC,其体内外活性、药代动力学稳定性、血浆半衰期均有明显提高[7]。
抗HER2-ADC的作用机制为:ADC通过受体介导的内吞作用进入细胞并转运至溶酶体,在溶酶体中被降解释放出pAF-AS269,结合到微管上,抑制微管的解聚,最终导致细胞死亡[24],因此,抗HER2-ADC能否与细胞表面HER2受体结合并内吞是决定其活性的重要因素。本研究在HCC1954细胞上证明了抗HER2-ADC可通过受体介导的内吞作用进入细胞。体外活性实验结果显示,抗HER2-ADC抑制HER2高表达的肿瘤细胞增殖,而对HER2低表达的肿瘤细胞无明显的生长抑制作用,说明了抗HER2-ADC对HER2受体的良好靶向性。曲妥珠单抗-美坦新偶联物也是一种以HER2受体为靶点的药物,但体外活性实验结果显示,其对HER2低表达的肿瘤细胞也有一定的抑制作用(表1)。曲妥珠单抗-美坦新偶联物中毒素小分子美坦新是通过异双功能的连接子4-(N-马来酰亚胺基甲基)环己烷-1-羧酸琥珀酰亚胺酯与曲妥珠单抗结合,虽然采用的是不可裂解的连接子,但是有研究表明4-(N-马来酰亚胺基甲基)环己烷-1-羧酸琥珀酰亚胺酯与美坦新之间的琥珀酰亚胺硫醚键可能不稳定,在氧化条件下会发生氧化反应,亚砜消除,进而释放美坦新的亚硫酸盐[25],另外,还有研究表明逆向迈克尔反应也有可能是毒素分子美坦新脱落的原因之一[26],这些脱落的毒性分子对HER2低表达的肿瘤细胞产生了抑制活性。而抗HER2-ADC对HER2低表达的肿瘤细胞无明显的抑制活性,间接表明抗HER2-ADC的稳定性更好。此外,小鼠体内活性实验中,抗HER2-ADC对HER2高表达的肿瘤细胞有明显的增殖抑制作用,且在相同剂量条件下抗HER2-ADC的药效优于曲妥珠单抗-美坦新偶联物。推测除了稳定性更好,抗HER2-ADC的药效优于曲妥珠单抗-美坦新偶联物的原因还有以下2个方面:首先,曲妥珠单抗-美坦新偶联物与抗HER2-ADC采用的毒性小分子与连接子均不相同,导致二者的代谢产物对HER2高表达的肿瘤细胞的增殖抑制作用产生差异;其次,临床前数据显示抗HER2-ADC在小鼠体内的半衰期比曲妥珠单抗-美坦新偶联物长(待发表), 因此,与曲妥珠单抗-美坦新偶联物相比,抗HER2-ADC表现出更优的药效。
综上所述,通过非天然氨基酸定点偶联技术获得的抗HER2-ADC,其作用机制和已上市的曲妥珠单抗-美坦新偶联物类似,其单克隆抗体具有特异性的靶向作用,对HER2高表达肿瘤细胞在体外细胞试验或小鼠体内试验都有很好的增殖抑制活性。后续我们会侧重于对抗HER2-ADC在动物体内代谢、毒理等情况进行评估,为抗HER2-ADC进入临床试验奠定数据基础,这对开发新的治疗HER2高表达肿瘤的方法具有重要的意义。