
炎症小体与非酒精性脂肪性肝炎
2019-10-27 08:24:52俞建顺陈芝芸
医学研究杂志 2019年12期
俞建顺 陈芝芸
非酒精性脂肪性肝病(non-alcoholic fatty liver disease,NAFLD)被认为是代谢综合征的一种表现。在全世界范围内,NAFLD的发生率约为25%,在亚洲,这一数字大约是27.4%[1]。大多数NAFLD患者不表现出任何症状,其中有约20%的患者进展为非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,NASH),NASH可进一步发展为肝硬化、肝细胞性肝癌等。时至今日,对于NASH的发病机制仍不十分确定,也缺乏有效的治疗手段。目前比较广为接受的是“二次打击”学说。第1次打击是肝细胞内脂质的累积,脂质的累积会导致肝脏损伤。饮食内或脂肪组织释放的多余游离脂肪酸以及肝脏内脂肪酸合成增加及分解减少均会导致脂质累积。第2次打击包括多种因素,例如细胞凋亡、坏死,氧化应激和脂质过氧化,促炎细胞因子的表达和线粒体功能失调,所有这些功能失常逐渐导致NAFLD进展为NASH和纤维化。尽管已经知道是第2次打击促进了NAFLD向NASH的发展,但目前为止还没有足够证据能够解释其中的分子机制。最新研究发现,炎症小体在NASH的发展中起到了重要作用,现对NASH和炎症小体的关系进行综述。
一、炎症小体概述
炎症小体是细胞内多蛋白复合物,在自身免疫性疾病、感染性和代谢性疾病中具有重要地位,是固有免疫的重要组成部分。目前为止,已证明多种炎症小体的存在,包括NLRP1、2、3、6、10、12、NLRC4和AIM2等。尽管在配体的识别、下游信号通路和生物功能等方面,各个炎症小体之间存在差异,但是几乎所有的炎症小体的核心功能都是激活含半胱氨酸的天冬氨酸蛋白水解酶-1(Casp1)。
大多数炎症小体以NOD样受体(nucleotide-binding oligomerization domain protein-like receptor,NLR)为核心,通过其识别病原体相关分子模式(pathogen-associated molecular patterns,PAMPs)或损伤相关分子模式(damage-associated molecular patterns,DAMPs),经含有CARD(caspase recruitment domain)的凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing a CARD,ASC)这一衔接蛋白使Casp1前体激活成为Casp1,活化的Casp1能够促进炎性细胞因子IL-1β和IL-18的成熟,从而发挥效应[2]。
炎症小体主要由固有免疫细胞表达,例如单核-吞噬细胞、树突状细胞和中性粒细胞等,在一些非免疫细胞中也有表达,例如角化细胞、肝细胞、肠上皮细胞等。具体来说,NLRP3炎症小体主要在粒-单核细胞系细胞内表达,在受到炎性刺激的时候,单核-吞噬细胞内NLRP3炎症小体的表达显著增加。另外还有报道T细胞、B细胞、口腔及生殖道上皮细胞、皮肤角质细胞和软骨细胞也都能表达NLRP3炎症小体[3]。NLRP6炎症小体主要在肾脏、肝脏、肺、小肠和大肠内表达,在肠道内,NLRP6主要由肠上皮细胞和结肠杯状细胞内表达,且和肠道微生物的组成有一定关系,另外,结肠内的成纤维细胞也可表达NLRP6[4]。NLRC4炎症小体在巨噬细胞、中性粒细胞和肠道上皮细胞内表达和激活[5]。AIM2炎症小体主要位于细胞质中,可在脾脏、小肠和外周血中检测到,在EV-A71感染的患者中枢神经系统中表达明显增加,且和多种肿瘤的发生有关,在结肠癌、前列腺癌和肝癌组织中均可检测到其异常表达[6]。
一般来说,模式识别受体(pattern recognition receptor,PRR)由病原体来源的抗原激活。NLR作为PRR的一种,同样如此。例如NLRP1炎症小体可由炭疽毒素激活,NLRP3炎症小体可由细菌来源的脂多糖(lipopolysaccharide,LPS)、病毒等激活,NLRC4可由沙门菌鞭毛蛋白激活,AIM2可由痘苗病毒激活。但是,除此之外,NLR同样也能由内源性的分子激活,例如NLRP1可被化学疗法激活,NLRP2可被ATP激活,NLRP3炎症小体可由多余ATP、葡萄糖、神经酰胺、活性氧簇、氧化LDL、尿酸和胆固醇结晶激活,AIM2可由DNA激活[7]。
研究证明,脂肪酸和LPS通过激活炎症小体在NASH的发展中起到了重要作用。食物中的饱和脂肪酸(棕榈酸)能够诱导Toll样受体(Toll-like receptor,TLR)2和TLR1形成二聚体,促进NLRP3的表达,刺激单核细胞释放IL-1β,从而导致外周组织的炎症[8]。棕榈酸在肝脏内能够诱导细胞凋亡和caspase-8的活化,从而导致危险信号的释放,并能增加肝细胞对脂多糖的敏感度,从而激活肝细胞内炎症小体,通过激活肝内单核细胞放大炎性反应,导致肝损的发生[9]。另一项研究显示,LPS的单独刺激足够引起肝脏NLRP3炎症小体活化通路下游炎性细胞因子的生成[10]。一般认为,革兰阴性菌感染后能够促进宿主细胞内NLRP3炎症小体(NLRP3、ASC和Casp1前体复合物)形成,其细胞壁内的LPS能够由细胞表面的受体TLR4和MD2共受体复合物识别,经一条TRIF衔接蛋白和1型干扰素参与的信号通路调控Casp11的表达,从而促进Casp1前体向Casp1转化[11]。
二、炎症小体与NASH
1.肝脏内炎症小体与NASH:肝脏炎症小体主要存在于库普弗细胞、肝实质细胞、肝内皮细胞及星状细胞内。Dixon等[12]研究发现使用蛋氨酸、胆碱缺乏饮食(MCDD)喂养诱导的小鼠NASH模型和使用普通饮食喂养的小鼠比较,肝内出现炎症小体激活和Casp1表达增加,并且伴有ASC mRNA表达水平升高;MCDD喂养的Casp1-/-小鼠和野型(wild type,wt)小鼠比较,多种炎性因子mRNA表达减少,说明Casp1的激活能够促进NASH进程中炎症的发展。同样,Csak等[9]的研究也证明了MCDD诱导的NASH的发生和肝内炎症小体相关蛋白(NLRP3、ASC和Casp1)表达的增加有关。此两项研究还发现,MCDD诱导的NASH小鼠肝内IL-1β水平显著升高,而IL-18却无明显变化[12]。
炎症小体的激活同样能够促进其他饮食诱导的NASH发展。使用高脂饮食(HFD)喂养的NLRP3-/-、ASC-/-、Casp1-/-小鼠和wt小鼠比较,肝细胞脂肪变度较轻,肥胖程度较轻,胰岛素敏感度较好,说明NLRP3炎症小体相关蛋白的缺失能够抑制HFD诱导的NASH的进展,且IL-1β能够促进NASH的发展[13]。而使用胆碱缺乏氨基酸(CDAA)饮食喂养的小鼠进行实验,结果同样支持肝内(主要为库普弗细胞)NLRP3炎症小体的激活会加重NASH进程和胰岛素抵抗这一结论,且该NASH的发展也和IL-1β有关[14]。
而在体外实验方面,Luo等[15]研究证明了通过抑制NLRC4炎症小体的活化、Casp1的激活及IL-1β的裂解,能够减轻PA诱导的HepG2细胞内的脂质蓄积和炎症,同样说明肝细胞内NLRC4炎症小体的激活会促进NASH的发展(图1)。
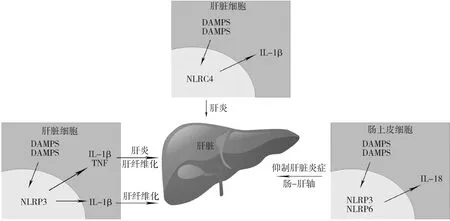
图1 各脏器炎症小体对NASH发生与发展的影响
2.肠道炎症小体与NASH:肠道菌群的失调和NASH的发生、发展有密切关系,而在这其中,炎症小体扮演了重要的角色。研究发现,炎症小体相关蛋白的缺陷(NLRP3-/-、NLRP6-/-、Casp1-/-小鼠)和上皮完整性及肠道炎症有关,后者可以导致肠道菌群失调及菌血症的发生,炎症小体的表达对肠道菌群的组成具有重塑作用[16, 17]。
Henao-Mejia等[18]研究发现,肠道内NLRP3和NLRP6炎症小体的缺陷可致肠道微生物发生改变,诱导结肠炎的发生,从而促进NASH的进展。肠上皮细胞内NLRP6和NLRP3炎症小体在维持肠道正常微生物生态平衡中起到了至关重要的作用,两者通过介导IL-18的激活维持肠道菌群的稳态,当其出现缺陷时,小鼠肠道内拟杆菌门细菌(属普雷沃菌科)和TM7菌门细菌增加,乳酸菌属细菌(属厚壁菌门)减少,这一菌群被称为致结肠炎菌群(colitogenic microbiota),这一菌群会诱导上皮细胞分泌CCL5,导致自发性和诱发性结肠炎的发生。而包括AIM2、NLRC4、NLRP10和NLRP12在内的其他炎症小体的缺陷均不会导致小鼠肠道致结肠炎菌群的发生。结肠炎的发生使肠黏膜屏障破坏,肠壁通透性增加,细菌产物(作为TLR4和TLR9激动剂,具体成分尚不清楚)透过肠壁进入门脉循环到达肝脏,激活肝内TLR4和TLR9。TLR4/9能够诱导肝内TNF-α的生成,从而加重MCDD诱导的NASH[18]。
同样,Pierantonelli等[19]使用高脂高糖饮食喂养诱导的NASH小鼠模型,也得出了类似的结论,高脂高糖饮食喂养的NLRP3-/-小鼠肠道内免疫应答的紊乱、抗菌肽表达的破坏、肠道通透性的增加以及肠道菌群的紊乱共同导致肠道菌群的易位,从而进一步加重NASH的进展。高脂高糖饮食喂养的NLRP3-/-小鼠肝脏损伤程度较野生型小鼠肝脏损伤程度加重,同时伴有革兰阴性菌(变形菌门及疣微菌门)数量的增加,这两种细菌能够降解肠道黏液,从而导致细菌易位的发生(图1)[19~21]。
综上所述,肠道内炎症小体(NLRP3及NLRP6炎症小体)的激活能够保护肠黏膜屏障,从而抑制NASH的发展,且该炎症小体激活通路下游的效应因子主要是IL-18。肝脏内炎症小体(主要是NLRP3炎症小体)的激活则会促进NASH进展,而该炎症小体激活通路下游的效应因子主要是IL-1β。
Chen等[22]研究发现,NLRP6-/-小鼠和wt小鼠比较,葡聚糖硫酸酯钠(dextran sulfate sodium,DSS)诱导的结肠炎明显加重,血清中和肝内IL-18水平的显著下降。Minicis等[23]在实验中同时测定了肠道内和肝内炎症小体相关蛋白的表达。该研究发现HFD喂养导致的肠道微生物紊乱能够促进胆管结扎(bile duct ligation,BDL)诱导的肝损伤和肝纤维化,而在该过程中,小鼠肝内TLR4和TLR9 mRNA的表达显著增加,炎症小体相关蛋白(包括NLRP3和ASC)的表达均显著升高,炎症小体活化的产物Casp1和IL-18同样有所升高。而在肠道内(尤其指肠上皮细胞)则恰好完全相反:HFD+BDL组小鼠盲肠中TLR4和TLR9基因及NLRP3、ASC、Casp1和IL-18的表达均显著下降。这似乎也说明了炎症小体在不同器官中起到了不同作用,对肝损伤起促进作用,但对肠道通透性和细菌易位起保护作用。然而,无论是肝内还是肠内,炎症小体功能的失调均会对NASH的发展产生影响。
需要指出的是,Henao-Mejia等[18]和Dixon等[12]的研究中同样使用了Casp1-/-小鼠,Casp1基因的敲除影响的是体内所有器官内Casp1的表达,不存在器官特异性,而两者在MCDD的喂养下却出现了相反的结果。经过比较,两个研究的实验过程并未存在明显不同,因此笔者大胆推测,这可能和实验小鼠初始状态下肠道菌群的差异有关,这也将成为课题组后期的工作之一。另外,Henao-Mejia等[18]的实验还发现,和wt小鼠比较,CD11c+骨髓细胞(Nlrp3KI;CD11c+-Cre)或肝细胞(Nlrp3KI;Albumin-Cre)特异性表达活化形式的NLRP3炎症小体的基因敲入小鼠在MCDD诱导的NASH严重程度上无明显区别,因此认为髓系细胞和肝细胞以外的细胞炎症小体功能失常是导致炎症小体缺陷小鼠NASH发展的决定因素。而上述多个研究表明肝组织内肝细胞以及库普弗细胞内的炎症小体在NASH的发展中起到了重要作用。有研究发现,髓系细胞特异性NLRP3炎症小体活化会导致严重的肝炎和纤维化[24]。同样,库普弗细胞中炎症小体的激活会导致胆固醇外流减少和自噬作用的失调,刺激胆固醇结晶的形成,导致肝炎的发生,这与Henao-Mejia等得出的结论存在冲突[25]。笔者推测,Henao-Mejia等[18]的研究使用的骨髓和肝细胞特异性表达NLRP3炎症小体的基因敲入小鼠体内存在持续的NLRP3炎症小体激活,而在MCDD的诱导下,wt小鼠骨髓细胞和肝细胞内NLRP3炎症小体被激活,同样出现了显著的肝炎表现,这可能反而更加能说明髓系细胞和肝细胞内NLRP3炎症小体的激活在NASH中起到的重要作用。另外还有研究发现NLRP6炎症小体对肠道微生物的组成并无影响,且不影响DSS诱导的结肠炎[26]。该研究认为,这可能是因为Henao-Mejia等[18]的研究未使用同窝小鼠,NLRP6-/-小鼠的致结肠炎菌群是随机获得的,和基因型无关,但具体原因有待于进一步研究。
三 炎症小体与NASH相关肝纤维化
目前关于炎症小体和NASH肝纤维化关系的研究主要集中在NLRP3炎症小体上。Wree等[27]的研究发现,NLRP3-/-小鼠可免于长期CDAA饮食喂养诱导的肝肿大、肝损伤及活化巨噬细胞的浸润。更重要的是,CDAA饮食喂养的NLRP3-/-小鼠肝纤维化显著减轻。而经过短期(4周)的CDAA饮食喂养,NLRP3基因敲入小鼠体内持续的NLRP3激活会诱导肝内严重的炎症(炎症评分增加,Casp1及IL-1β前体mRNA水平升高)、巨噬细胞浸润、HSC的激活和明显的肝纤维化发生,而同样饮食喂养的WT小鼠则只有轻度的单纯性肝脏脂肪变性。这些结果共同说明NLRP3炎症小体的激活参与NAFLD肝纤维化的发生、发展。
另外,该研究还对NAFLD疾病谱不同阶段(主要分为NASH患者及非NASH患者)的患者肝组织进行研究,结果发现NASH患者肝组织内NLRP3炎症小体相关蛋白(NLRP3、IL-1β前体、IL-18前体)表达显著高于非NASH患者,且I型胶原A1(collagen type I alpha 1,COL1A1)和IL-1β前体的mRNA两者的水平具有相关性。胶原沉积是肝纤维化的重要表现,这同样支持NLRP3炎症小体的激活在NAFLD患者肝纤维化进展中起到了重要作用。
研究认为,肝细胞及肝非实质细胞内NLRP3炎症小体信号通路激活后,Casp1活化,从而诱导肝细胞焦亡的发生,HSC内NLRP3信号通路的激活则会导致胶原沉积的加剧,而这两者均会导致肝纤维化的发生[24,28,29]。NLRP3炎症小体激活导致肝纤维化,其下游的效应因子IL-1β起到了重要作用,而有研究使用阿那白滞素(一种IL-1受体拮抗剂)治疗NLRP3炎症小体激活小鼠,其肝脏炎症可得到部分缓解,但HSC的活化及肝纤维化则无明显影响,这说明NLRP3炎症小体的激活导致肝纤维化发生的信号通路下游除IL-1β外还有其他效应因子,而这其中可能就包括TNF信号通路[24,30]。
四、展 望
社会的发展、生活水平的提高,带来的是日益严重的肥胖问题,也正因为如此,非酒精性脂肪性肝病的发生率逐年升高。作为代谢综合征的一部分,NAFLD/NASH和糖尿病、高脂血症等疾病往往是互相伴随的,且NAFLD/NASH已成为隐源性肝硬化的最主要原因。目前为止,对于NAFLD/NASH的治疗仍然主要局限在生活方式的改变、减重等发面,药物治疗的效果并不确切,因此,进一步明确本病的发病机制,寻找治疗靶点,开发新的药物迫在眉睫。综上所述,炎症小体直接参与到NAFLD疾病谱中单纯性脂肪变向NASH的转变及进一步肝纤维化的发生中,在NASH的发生与发展中起到了重要的作用。但是,目前研究基本集中在NLRP3和NLRP6炎症小体,且尚处于比较初级的阶段,研究并不是十分彻底,甚至在一些问题上许多研究之间还产生了明显的分歧,尤其是在不同器官内炎症小体对NASH的影响,部分研究甚至出现了相反的结果。但是,相信随着对炎症小体进一步深入研究,对NASH的认识会越来越深入。
猜你喜欢
中老年保健(2022年2期)2022-08-24 03:20:50
昆明医科大学学报(2022年2期)2022-03-29 00:52:18
科学(2020年4期)2020-11-26 08:27:06
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:43
癌变·畸变·突变(2016年3期)2016-02-27 06:15:36
哈尔滨医药(2015年4期)2015-12-01 03:57:54
动物营养学报(2015年10期)2015-12-01 02:26:20
西南军医(2015年3期)2015-04-23 07:28:32
国际心血管病杂志(2015年5期)2015-02-27 12:11:34
现代检验医学杂志(2015年4期)2015-02-06 02:02:11
