
高效液相色谱-荧光检测法测定食品中维生素K2的含量
2019-10-25 02:37
食品工业科技 2019年19期
(深圳市计量质量检测研究院,国家营养食品质量监督检验中心(广东),广东深圳 518131)
天然维生素K(Vitamin K,VK)是一类基本结构为2-甲基-1,4-萘醌的脂溶性维生素,参与人体骨代谢和细胞生长,是肝内合成凝血酶原的必需物质,具有止血功能,当体内缺乏时可造成凝血障碍[1]。根据其侧链结构的不同,天然维生素K可分为维生素K1(Vitamin K1,VK1)、维生素K2(Vitamin K2,VK2)两类。维生素K1亦称叶绿醌,在母环C-3位置上有一个植烷取代基,主要存在于绿色植物中。维生素K2亦称甲萘醌,是系列化合物,根据母环C-3位置上异戊二烯单元个数的不同,分为14种,以MK-n表示(图1),其中n指异戊二烯单元的个数[2],维生素K2主要由微生物代谢产生,少量存在于肉类、动物内脏、鸡蛋、乳制品、发酵性食品中,在纳豆中较丰富[3]。维生素K2是唯一具有生物活性的维生素K,其中以四烯甲萘醌(Menatetrenone 4,MK-4)、七烯甲萘醌(Menatetrenone 7,MK-7)最具代表性[4]。然而目前国内主要以维生素K1作为添加剂添加到食品中,以补充人体内维生素K的不足,维生素K2发展仍属起步阶段。但维生素K1需在肝脏内转化为维生素K2才能被人体吸收利用,其在体内的生物利用率仅为维生素K2的二分之一[5]。
随着其药用价值的突显,维生素K2已然成为国际上新一代预防和治疗骨质疏松的保健食品[6]。1995年,日本首次将维生素K2作为治疗骨质疏松药物应用[2]。2008年,美国食品药品管理局(Food and Drug Administration,FDA)将纳豆枯草芽孢杆菌发酵的MK-7列入安全类食品添加剂[7]。2009年,欧盟(European Union,EU)通过2009/345/EC决议,批准以纳豆枯草芽孢杆菌发酵的维生素K2作为一种新型食品配料投放市场[8]。我国台湾地区也将维生素K2作为第八类“营养添加剂”添加到食品中[7]。2016年,国家卫生和计划生育委员会在《关于海藻酸钙等食品添加剂新品种的公告(2016年第8号)》中将维生素K2(发酵法)列入食品营养强化剂新品种[9]。
目前,国内尚未有食品中维生素K2检测的相关标准,相关检测方法报道也较少,主要包括高效液相色谱-紫外检测法[10-13]、毛细管电泳法[14]、高效液相色谱-质谱法[15]等。维生素K2在食品中含量极少,且易受强酸、氧化剂、碱及光的作用而分解。现有的高效液相色谱-紫外检测法前处理采取的方法有直接萃取法和薄层纯化法;直接萃取法面对的样品主要是原料药、纳豆提取物等基质较为干净、含量较高的样品,而食品类别繁多、基质复杂、含量较低,直接萃取不完全、杂质干扰大;薄层纯化法前处理复杂,无法满足高通量检测;紫外检测器的选择性和灵敏度不令人满意,无法满足食物中维生素K2的检测需求[10-13];毛细管电泳法步骤繁琐、重现性差;高效液相色谱-质谱法灵敏度较高,但仪器复杂、价格昂贵,不易推广。维生素K的不饱和酮结构共轭体系较小,不能产生荧光,但能通过衍生法使维生素K同系物的母核萘琨环产生荧光,以提高其在高效液相色谱荧光检测器上的响应值,从而达到检测目的[16]。王琳等[17]用SnCl2的乙醇溶液将维生素K2荧光化,通过荧光检测器成功测得人体血液中MK-4的含量。韩一秀等[18]用L-半胱氨酸将维生素K3还原为有荧光的甲萘酚,建立了用水溶液测定药物制剂中维生素K3含量的新的荧光分析方法。
以上方法均是在前处理过程中将维生素K荧光化,本文参考AOAC Official Method 999.15[16],针对食品中MK-4、MK-7,采用柱后衍生反应使非荧光化合物维生素K2产生荧光,并结合荧光检测器进行检测,从而建立食物中MK-4、MK-7高效、准确、快速、灵敏的同时分离检测技术,以期为食品中维生素K2的检测、应用和监管提供技术支撑。
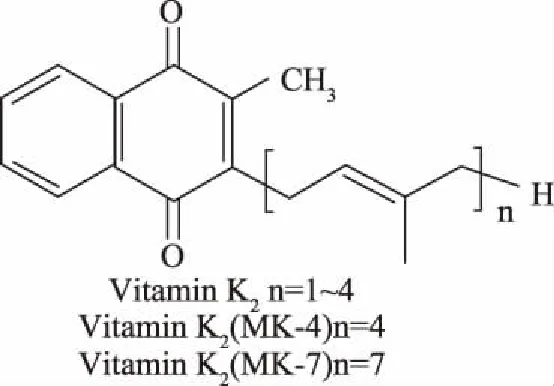
图1 维生素K2化学结构式Fig.1 Chemical structure of vitamin K2
1 材料与方法
1.1 材料与仪器
猪肉、猪肝、纯牛奶、酸奶、黄油、鸡蛋、豆豉、纳豆 市售;MK-4标准品 美国Supelco公司;MK-7标准品 美国ChromaDex公司;脂肪酶(酶活力≥700 U/mg) 美国Sigma公司;氢氧化钾、冰乙酸、氯化锌、无水乙酸钠、氯化钠、无水硫酸钠 分析纯,广州化学试剂厂;无水乙醇 分析纯,广州光华科技股份有限公司;甲醇、正己烷、二氯甲烷 色谱纯,德国Merck公司。
ThermoUltimate 3000液相色谱仪(配荧光检测器) 美国Thermo公司;BSA224S-CW型电子分析天平北京赛多利斯公司;MS3涡旋仪、KS4000ic空气控温摇床 德国IKA公司;Hei-VAP真空旋转蒸发仪 德国海道夫公司;N-EVAP112氮吹仪 中国路易公司;Agilent ZORBAX SB-C18色谱柱(150 mm×4.6 mm,5 μm) 美国Agilent公司;柱后锌粉还原柱(35 mm×4.6 mm,填料为锌粉,平均粒径70 μm) 上海安谱实验科技股份有限公司。
1.2 实验方法
1.2.1 标准品溶液制备 单标储备液:分别准确称取MK-4、MK-7标准品,用甲醇溶解并定容,配制质量浓度均为2000 mg/L的单标储备液,于-20 ℃冰箱避光保存。混合标准中间溶液:分别吸取MK-4、MK-7标准储备液1.00 mL于100 mL棕色容量瓶中,用甲醇定容至刻度,摇匀,于-20 ℃冰箱避光保存。
1.2.2 样品制备 称取上述固体样品约2 g、液体样品约10 g(精确至0.0001 g)于50 mL离心管中,加入约1 g脂肪酶、10 mL去离子水,振荡溶解,于(37±5) ℃空气控温摇床中酶解3 h。取出酶解好的试样,依次加入5 mL 无水乙醇、2 mL 50%氢氧化钾溶液,振荡混匀,使其皂化完全。加入30 mL 正己烷,充分振摇,于5000 r/min下离心3 min,取上清液于另一50 mL离心管中,加入10 mL饱和氯化钠溶液,振荡水洗,于5000 r/min下离心3 min,取上清液于100 mL旋蒸瓶中。重复操作1次,合并有机相,将有机相经过无水硫酸钠脱水过滤到旋蒸瓶中,在旋转蒸发仪上于(40±2) ℃蒸发至近干,氮气吹干,准确加入2.00 mL甲醇复溶,0.22 μm滤膜过滤,取滤液备用。
1.2.3 色谱条件 色谱柱:Agilent ZORBAX SB-C18色谱柱(150 mm×4.6 mm,5 μm);柱后锌粉还原柱:35 mm×4.6 mm,连接于色谱柱之后,检测器之前;柱温:25 ℃;流速:1.5 mL/min;进样量:10 μL;检测波长:激发波长243 nm,发射波长为430 nm;流动相:甲醇(含有二氯甲烷10%、冰醋酸0.03%、氯化锌1.5 g/L、无水乙酸钠0.5 g/L)。
1.2.4 维生素K2含量的计算 按下列公式计算样品中MK-4或MK-7含量:
X=c×V×d×100/m
式中,X为样品中MK-4或MK-7含量(μg/100 g),c为待测液中MK-4或MK-7的质量浓度(μg/mL),V为样品定容体积(mL),d为稀释倍数,m为样品称样量(g)。
1.3 数据处理
通过与仪器配套的Chromeleon 7工作站软件完成数据的采集与处理。
2 结果与讨论
2.1 酶解条件的选择
维生素K2是脂溶性维生素,在食品中与脂质共存。脂肪酶可有效降解食品中脂肪,但酶的用量及酶解时间对维生素K2的提取有一定影响。本文以鸡蛋为样品,考察了不同用量(0.5、1.0、1.5 g)的酶(见表1)和不同酶解时间(1、2、3、4、5 h)下样品的测定结果(见表1)。结果表明,在相同酶解时间条件下(5 h),样品在酶用量为1.0 g时可酶解完全。且实验过程发现,酶的用量与样品萃取过程中乳化现象有关,酶用量的增加可有效消除乳化现象的产生,有利于萃取过程中的液液分层。结合经济考虑,本文选取1.0 g酶用量。在相同酶用量条件下(酶用量1.0 g),酶解1、2 h结果偏低,酶解时间不足导致样品酶解不完全。酶解3、4、5 h样品测定结果趋于稳定,结合考虑时间效率问题,本文选取酶解时间为3 h。

表1 酶解条件的比较Table 1 Comparison of enzyme solution conditions
2.2 皂化条件的选择
食品中维生素K2常与脂质共存,极少以游离态形式存在,且食品中其他类脂类化合物的存在也是维生素K2检测的干扰来源,因此需要对食品进行皂化,破坏维生素K2外的包被物质,使维生素K2游离出来。在皂化反应中,碱的作用是将酶解后的脂肪转化成脂肪酸盐,将维生素K2释放出来,以便于萃取。由于维生素K2对碱敏感[2],本文选用鸡蛋为样品,选择50%氢氧化钾溶液为碱液,对碱的用量(1、2、3、4、5 mL)进行了考察(见表2)。结果表明,当碱用量1 mL时,结果偏低,这可能与碱用量不够、样品皂化不完全有关。碱用量2 mL可以满足实验需求。碱用量3、4、5 mL结果逐渐递减,可能与碱液含量过高,维生素K2被碱破坏有关。综合考量,本文选择使用2 mL 50%氢氧化钾溶液。

表2 皂化条件的比较Table 2 Comparison of saponification conditions
2.3 流动相的选择
考察甲醇(A,见图2)、甲醇(含二氯甲烷5%)(B,见图2)、甲醇(含二氯甲烷10%)(C,见图2)、甲醇(含二氯甲烷15%)(D,见图2)(均含有冰醋酸0.03%、氯化锌1.5 g/L、无水乙酸钠0.5 g/L)条件下目标峰分离度及出峰时间。结果表明,MK-4、MK-7在所有流动相条件下均可有效分离,流动相中添加二氯甲烷可有效改善峰形。随着二氯甲烷比例的增加,目标峰出峰时间加快。考虑到二氯甲烷对仪器管路具有腐蚀作用,本文选择使用甲醇(含有二氯甲烷10%、冰醋酸0.03%、氯化锌1.5 g/L、无水乙酸钠0.5 g/L)为流动相。在该条件下,MK-4的出峰时间为3.560 min,MK-7的出峰时间为11.332 min,表明该色谱条件有效可行(见图2,C)。

图2 MK-4、MK-7标准品色谱图Fig.2 Standard chromatogram of MK-4,MK-7
2.4 线性关系、检出限及定量限
将混合标准中间溶液逐级稀释成浓度为0.02~2.00 μg/mL的系列工作液,在1.2.3的色谱条件下检测,根据各峰面积(y)对质量浓度(x)进行线性回归,得到MK-4、MK-7的标准曲线方程、线性范围和相关系数。MK-4标准工作溶液线性回归方程为:y=1.37×106x-4.49×103,相关系数r=1.0000。MK-7标准工作溶液线性回归方程为:y=6.01×105x-2.58×103,相关系数r=1.0000。检出限和定量限分别以3倍和10倍信噪比(S/N)计,结果表明,当取样量为2 g,定容2 mL时,MK-4的检出限为0.01 μg/100 g,定量限为0.04 μg/100 g;MK-7的检出限为0.06 μg/100 g,定量限为0.2 μg/100 g。当取样量为10 g,定容2 mL时,MK-4的检出限为0.002 μg/100 g,定量限为0.008 μg/100 g;MK-7的检出限为0.012 μg/100 g,定量限为0.04 μg/100 g。

表3 鸡蛋和豆豉中维生素K2的加标回收率和相对标准偏差(n=3)Table 3 Spiked recoveries and RSDs of VK2 in eggs and fermented soya beans(n=3)
2.5 回收率与精密度
根据2.6实际样品的测定结果,选取鸡蛋、豆豉做加标实验考察方法准确度。鸡蛋中含有MK-4,不含MK-7,向鸡蛋中添加0.5、1、2倍含量的MK-4,选取标准曲线低、中、高含量添加MK-7。豆豉中不含MK-4,含有MK-7,向豆豉中添加标准曲线低、中、高含量的MK-4,按0.5、1、2倍含量添加MK-7。每个水平平行3份(见表3)。MK-4的加标回收率在85.8%~94.1%之间,MK-7的加标回收率在80.6%~95.5%之间,精密度RSD均不超过3.56%,结果表明该方法回收率良好,精密度较高,实验结果准确。
2.6 稳定性
取上述加标样液,于-20 ℃避光分别保存0、4、8、12和24 h,考察样品中MK-4、MK-7的稳定性(见表4)。结果发现,在24 h内,MK-4、MK-7含量的相对标准偏差(RSD)均不超过8.69%,说明MK-4、MK-7的稳定性好。

表4 提取液存放时间对维生素K2含量的影响(n=3)Table 4 Effect of different storage time on the contents of VK2(n=3)

表5 8种食品中维生素K2的含量(μg/100 g,n=6)Table 5 Contents of VK2 in the 8 kinds of food(μg/100 g,n=6)
注:ND表示未检测到。
2.7 实际样品的测定
本方法选取猪肉、猪肝、纯牛奶、酸奶、黄油、鸡蛋、豆豉、纳豆8种样品进行检测(见表5)。结果表明,猪肉、纯牛奶、酸奶、黄油、鸡蛋中仅含有MK-4,其中纯牛奶中含量最低,仅为(0.34±0.02) μg/100 g,鸡蛋中含量最高,达(25.38±0.85) μg/100 g。猪肝、豆豉、纳豆中仅含MK-7,其中猪肝含量较低,为(25.45±0.83) μg/100 g,纳豆中含量最为丰富,高达(923.20±11.45) μg/100 g。本文所测样品中维生素K2含量与Schurgers等[3]、Peter等[19]的研究结果相符。
3 结论
本文建立了高效液相色谱-荧光检测法同时分离检测食品中2种维生素K2(MK-4、MK-7)的分析方法。MK-4、MK-7在15 min内分离出峰。2种维生素K2在各自的线性范围内关系良好,相关系数r=1.0000。检出限和定量限分别以3倍和10倍信噪比(S/N)计,当取样量为2 g,定容2 mL时,MK-4的检出限为0.01 μg/100 g,定量限为0.04 μg/100 g;MK-7的检出限为0.06 μg/100 g,定量限为0.2 μg/100 g。当取样量为10 g,定容2 mL时,MK-4的检出限为0.002 μg/100 g,定量限为0.008 μg/100 g;MK-7的检出限为0.012 μg/100 g,定量限为0.04 μg/100 g,精密度(RSD)均不超过3.56%,回收率为80.6%~95.5%。方法对猪肉、猪肝、纯牛奶、酸奶、黄油、鸡蛋、豆豉、纳豆8种样品进行检测,检测结果表明,猪肉、纯牛奶、酸奶、黄油、鸡蛋中仅含有MK-4,其中纯牛奶中含量最低,仅为(0.34±0.02) μg/100 g,鸡蛋中含量最高,达(25.38±0.85) μg/100 g。猪肝、豆豉、纳豆中仅含MK-7,其中猪肝含量较低,为(25.45±0.83) μg/100 g,纳豆中含量最为丰富,高达(923.20±11.45) μg/100 g。综上所述,该方法线性范围广,精密度和准确度良好,分析时间快,可进一步用于食品中维生素K2的研究。
猜你喜欢
钛工业进展(2022年6期)2023-01-13
化工设计通讯(2022年10期)2022-12-31
波谱学杂志(2022年2期)2022-06-14
化工设计通讯(2020年11期)2020-01-12
中国化肥信息(2019年6期)2019-08-27
人间(2015年11期)2016-01-09
质量安全与检验检测(2012年6期)2012-09-17
