
TCGA数据库中不同种族肝细胞癌相关lncRNAs的筛选及功能预测
2019-10-24 08:28马佳康任凯凯李雨濛孙金旗
医学研究生学报 2019年10期
马佳康,任凯凯,李雨濛,李 南,张 玲,王 健,孙金旗,周 博,马 军
0 引 言
原发性肝细胞癌是常见的恶性肿瘤[1],据欧美国家统计显示,从2010年开始,男性肝细胞癌发病率处于平台期,而女性肝细胞癌发病率却在迅速增加;通过加强肝炎病毒筛检和研发新的抗病毒药物,可明显降低肝细胞癌的风险[2]。因此,我们需要进一步探索其发病机制,找到更有效的诊断标记和治疗靶点。
数据显示人类基因组中90%以上的序列可以被转录,但只有1%~2%的序列可以编码蛋白质。根据其转录本的长度可以分为<200 个核苷酸的小RNA,和>200 个核苷酸的长RNA(lncRNAs)。近年来发现lncRNAs可能在肿瘤发生发展中发挥重要作用,但对于其生物学功能目前仅有少部分被证实。lncRNAs 具有多种功能,如诱饵、支架,指导信号分子参与许多重要的生物过程,如染色体沉默、基因组印记、染色质修饰、转录调节和miRNAs 功能调节[3-4]。其中,lncRNAs 通过竞争性內源RNA(ceRNA)机制调控基因表达也已经被证实[5-6]。肝细胞癌相关lncRNAs 的研究,将为其预防和控制提供新的思路和方法[7]。目前,已发现数十种和肝细胞癌相关的lncRNAs,如H19、HULC、MALAT-1、HOTAIR和HOTTIP 等[5,8-13]。本研究旨在分析TCGA 数据库中白色人种、黑色人种和黄色人种肝细胞癌共有和特有lncRNAs差异谱并预测其功能和调控机制。
1 资料与方法
1.1 数据下载和差异基因筛选从TCGA 数据库共下载349例肝细胞癌患者的临床资料和转录组数据。另外下载相应病例的miRNAs 测序数据。通过R 语言edgeR 包对mRNAs、lncRNAs 及miRNAs 的基因样本分别进行差异分析,标准为|log2FC|>2 且FDR<0.05[14]。
1.2 lncRNAs 筛选和功能分析通过对白色人种、黑色人种和黄色人种肝细胞癌患者的癌和癌旁组织进行差异分析得到各自的差异的lncRNAs,并比较3 个人种差异lncRNAs 基因谱情况,采用韦恩图找出3个人种共有的差异lncRNAs。
1.2.1 ceRNA 网络构建用肝细胞癌中差异的lncRNAs、mRNAs 和miRNAs,通过数据库miRcode、starBase、miRDB、miRTarBase 和TargetScan 建立lncRNAs-miRNAs-mRNAs的关系对,通过Cytoscape 软件从而构建ceRNA网络。
1.2.2 lncRNAs 预后分析以本组所有肝细胞癌患者的临床数据为基础,对3 个人种共同差异的lncRNAs 进行Cox 回归分析,按P<0.05 或P<0.01 选取lncRNAs,最终得到和肝细胞癌生存相关的lncRNAs组合。
1.2.3 lncRNAs 靶基因预测和功能预测对多因素Cox 分析得到的lncRNAs,通过MEM-Multi Experiment Matrix(https://biit.cs.ut.ee/mem/)做靶基因预测(pearson 相关)。对lncRNAs 的靶基因通过R语言的clusterProfiler 包,进行GO 及KEGG 分析,并对结果进行可视化;使用Cytoscape 软件的iRegulon插件预测分析调控这些靶基因的转录因子;以FDR<0.001,富集分数>3.0 以及Targets>10 为筛选条件,来筛选相应靶基因的转录因子。
1.3 特有的lncRNAs 相关分析分别对白色人种和黄色人种特有的lncRNAs 进行多因素Cox 分析(黑色人种病例数太少不做分析),对靶基因分别进行GO、KEGG分析和可视化;iRegulon插件预测相关转录因子。
2 结 果
2.1 差异lncRNAs 筛选白色人种筛选出的差异lncRNAs 上调989(94%),下调60(6%);黄色人种上调152(65%),下调83(35%);黑色人种上调301(77%),下调88(23%)。见图1。
2.2 3 个人种共有lncRNAs 筛选、生存分析和功能预测通过韦恩图取交集发现,3 个人种共有的差异表达的lncRNAs 有49 个,白色人种和黑色人种重叠基因21.5%,白色人种和黄色人种、黑色人种与黄色人种差别较大,其差异lncRNAs 重叠率仅为7.8%和5.8%,见图2a。对49 个lncRNAs 进行靶基因预测,并建立了相关ceRNA 网络图,见图2b,其中lncRNA HOTTIP、hsa-mir-424、hsa-mir-373、hsa-mir-182和hsa-mir-519d是重要的节点基因。
49个差异的lncRNAs行多因素Cox分析,建立了预 后 诊 断 组 合,含 有7 个lncRNAs(AC010280.2、AC099508.2、LINC02475、LINC02313、AC138356.1、LINC01224 以及LINC02561),组合能很好判断患者的预后(AUC=0.709),其中LINC01224可预测到相应的靶基因。对LINC01224预测的靶基因行GO富集发现,其功能和DNA复制、转位、基因表达调控、表观遗传、miRNAs 基因沉默及RNA 转录后基因沉默等有关。KEGG 分析显示,LINC01224 和细胞周期调控、DNA 复制相关。见图3。其靶基因的转录因子预测发现,LINC01224 和TAF7、E2F4、HDAC2、NANOG、SOX5、E2F1、DEAF1、FOXN4、MAFB、ZNF513、TFDP3、ATF2、NKX2-3和EBF1相关,见图4。
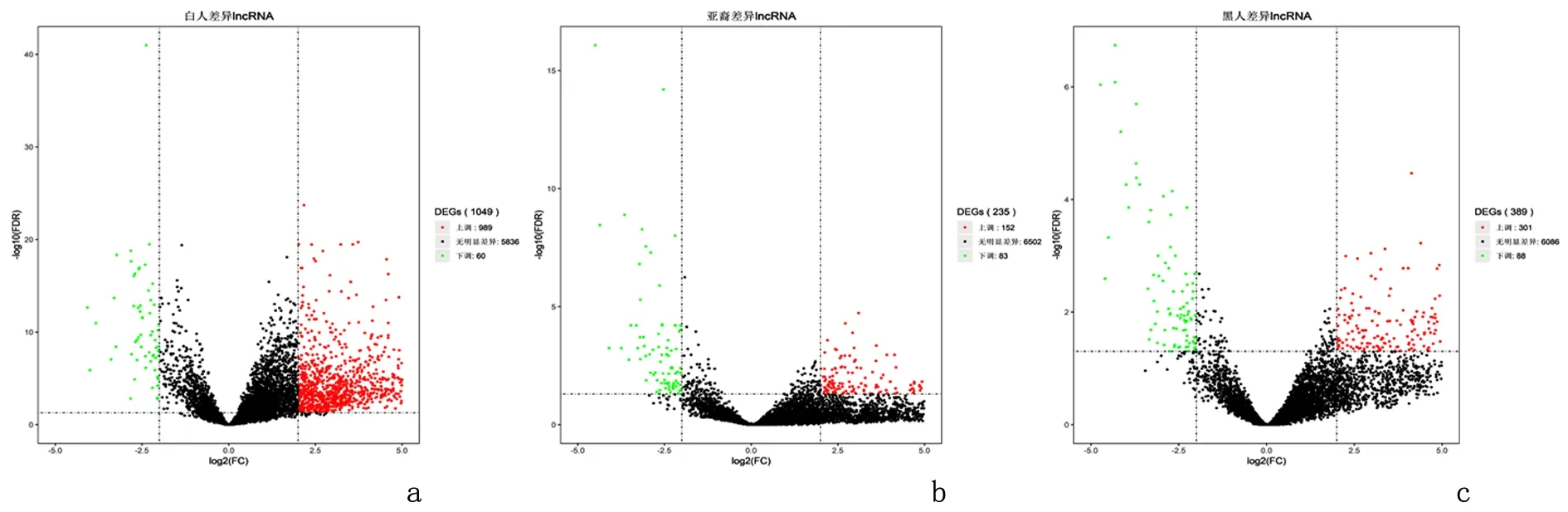
图1 不同人种肝细胞癌差异lncRNAs筛选Figure 1 Screening of differentially expressed lncRNAs in different human races in the TCGA database
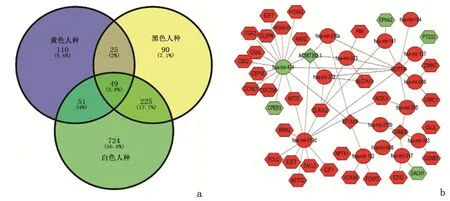
图2 共有的差异lncRNAs筛选和ceRNA调控网络的构建Figure 2 Screening of shared differentially expressed lncRNAs and construction of the ceRNA regulatory network
2.3 白色人种和黄色人种特有的lncRNAs相关分析维恩图分析发现,白色人种有724个特有lncRNAs。Cox生存相关分析后发现含11 个lncRNAs 的预后组合(AC008892.1、AC015722.2、AC073987.1、AC090568.2、AC136188.1、AL356215.1、AL591501.1、GACAT3、LINC02327、PLUT以及Z93403.1),组合能很好判断患者的预后(AUC=0.767),但在11个基因中未预测到靶基因。黄色人种特有的lncRNAs 110个,Cox分析后筛选出含6 个lncRNAs 的预后基因组合(AC006252.1、AC009005.1、AC025048.4、AC093609.1、AC126118.1以及AL024498.1),组合能很好判断患者的预后(AUC=0.846)。黄色人种肝细胞癌生存相关的6个lncRNAs中,lncRNAs AC126118.1和AC093609.1可在现有MEM数据库中找到靶基因,分别预测到34、37个靶基因。
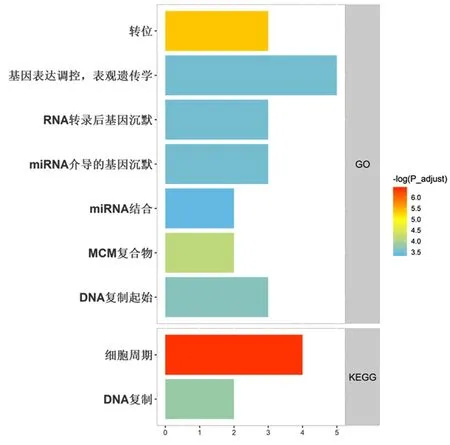
图3 LINC01224的GO、KEGG分析Figure 3 GO and KEGG analyses of LINC01224
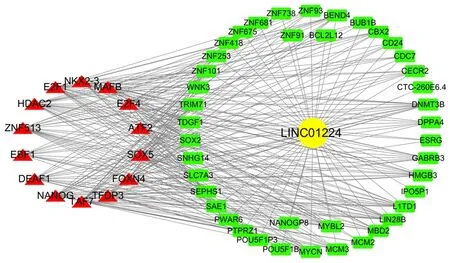
图4 LINC01224转录因子预测Figure 4 Transcription factor prediction analysis of LINC01224
AC093609.1 的靶基因通过GO 富集后结果显示,其功能和离子通道活性如钙通道活性、被动跨膜转运蛋白活性有关;KEGG 结果显示其参与心肌病、心肌细胞肾上腺素能等信号通路的调控。预测得到AC093609.1 的转录因子为GLI2、FLI1 和HOXB7。AC126118.1 的靶基因通过GO 富集后发现,其靶基因和多种代谢功能相关,如羧酸分解代谢过程、脂肪酸β-氧化、脂肪酸分解代谢等;也与酶活性有关,如酰基辅酶A 脱氢酶活性、氧化还原酶活性、ATP 酶活性等,还与跨膜运动相关;KEGG 结果也显示其参与脂肪酸降解、ABC 转运蛋白、氨基酸代谢等信号通路。AC126118.1 靶基因相关的转录因子有ESR1、ELK1、FOS、MYBL1、JDP2、EGR1、DEAF1 和POLR2A,见图5、图6。
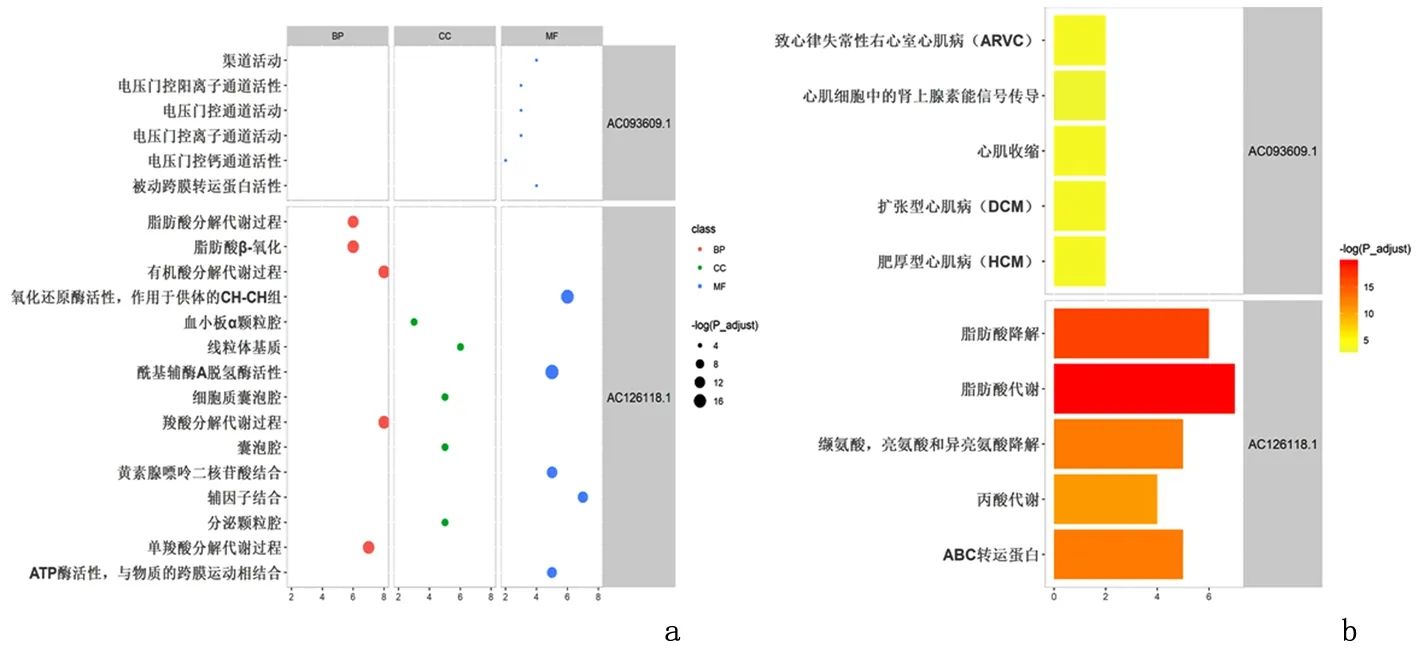
图5 lncRNAs AC126118.1和AC093609.1的功能预测分析Figure 5 Functional prediction analysis of the lncRNAs AC126118.1 and AC093609.1
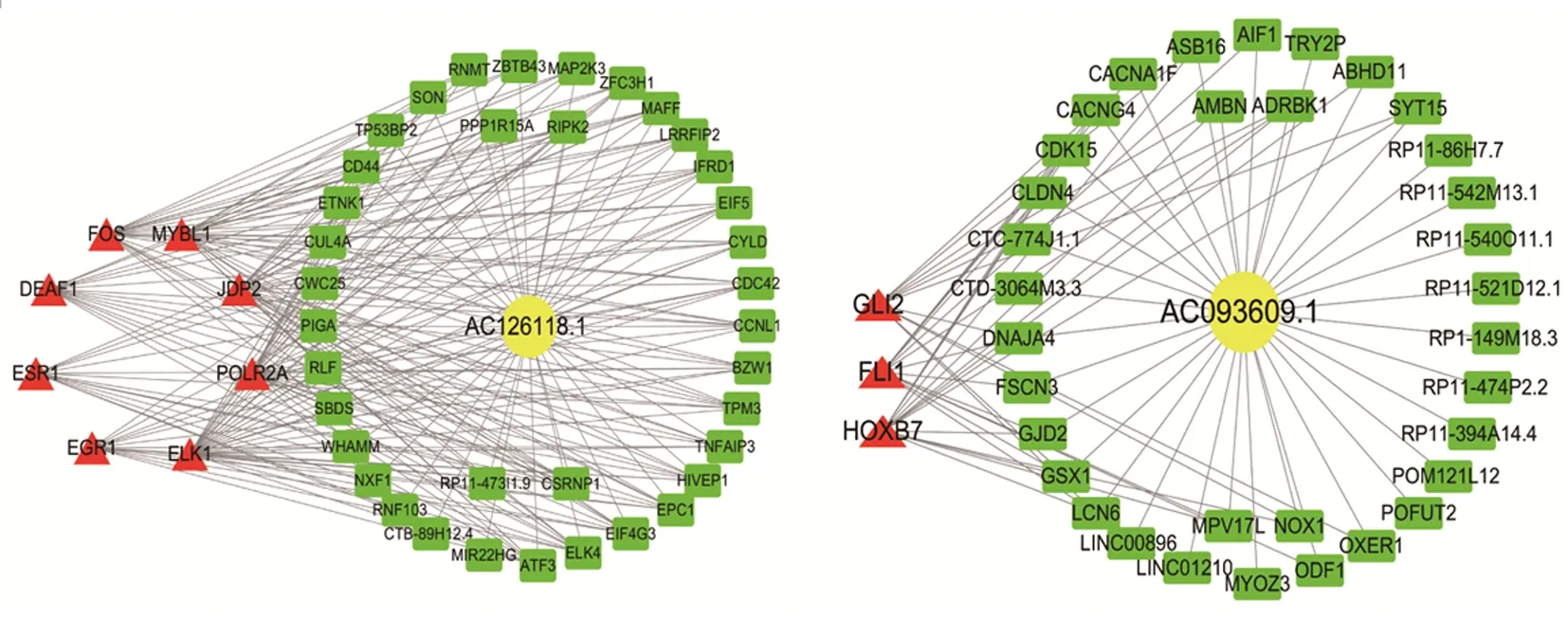
图6 lncRNAs AC126118.1和AC093609.1的转录因子预测Figure 6 Transcription factor prediction analysis of the lncRNAs AC126118.1 and AC093609.1
3 讨 论
本研究首先分析了TCGA 数据库中的HCC 的lncRNAs表达数据,发现在3个人种中lncRNAs表达谱差异较大,其中白色人种和黑色人种的lncRNAs表达谱相似性更高一些,而黄色人种和白色人种以及黑色人种的差异较大。其中有49 个共同表达的差异lncRNAs 可以作为HCC 的共有诊疗标志物。为了进一步研究HCC 的发生机制和判断这些指标在诊疗中的价值,对49 个lncRNAs 进一步行生物信息学分析,利用现有数据库信息,成功构建了ceRNA 网络,其中以hsa-mir-424,hsa-mir-519d,hsa-mir-182,hsa-mir-373 为中心,组成了lncRNAs 和mRNAs的调控网络,为今后进一步研究HCC 的表观遗传学调控提供了一定线索。生存相关的基因常常在肿瘤发生发展中起着重要作用,因此本研究对共有lncRNAs 进行Cox 回归分析,发现了含有7 个lncRNAs的组合对HCC具有重要的预后价值。由于lncRNAs的研究数据尚少,本研究只发现LINC01224 有相应的功能和信号通路数据。本研究对LINC01224进行GO、KEGG 和转录因子网络分析,这为LINC01224的调控机制的研究奠定了基础。
由于各人种存在一定的遗传差异,因此在HCC发生发展中,lncRNAs 的表达谱有明显差异,其中起重要调控作用的lncRNAs 也不同,在Venn 图分析中,有更多的lncRNAs 表现为人种特异性。通过对这些人种特异lncRNAs 行COX 回归分析,我们分别找出了与白色人种(11个)和黄色人种(6个)预后相关的lncRNAs组合,由于黑色人种例数较少,所以未对其进行生存分析。2 个人种的生存相关lncRNAs组合中无重叠组分,说明HCC 相关的关键预后lncRNAs 存在差异。为了对白色人种和黄色人种预后相关的差异lncRNAs 作进一步研究,我们对其进行靶基因预测,在MEM 数据中,未发现与白色人种HCC 预后相关的lncRNAs 的靶基因,说明对这些IncRNA 的功能目前未知。在黄色人种预后相关的lncRNAs 中,可 以 找 到lncRNAs AC126118.1 和AC093609.1 靶基因,通过GO、KEGG 和转录因子预测分析,有助于今后的功能研究。
TCGA 数据库拥有海量的信息,包含转录组、表达谱、基因突变、单核苷酸多态性及甲基化等信息。通过生物信息学对现有的数据库进行挖掘,我们可以整合分析肿瘤相关异常信息,为进一步的功能验证和机制研究奠定基础。已发表的关于HCC 的数据挖掘报道,主要针对GEO 数据,因为GEO 数据库建立较早[6,15],GEO 数据主要是芯片数据,病例数较少,临床信息不全面,影响了数据挖掘结果的质量。在已发表的TCGA 数据挖掘文献中,关于lncRNAs相关的整合分析[9,16],未考虑人种不同和lncRNAs表达的相关性。
本研究针对人种不同,分析了白色人种、黄色人种和黑色人种共有及特有的相关lncRNAs差异表达谱,并发现了一些不同人种共有和特有的和预后相关的重要lncRNAs,为今后深入的了解肝细胞癌的发病机制、寻找新的诊疗靶点提供了重要依据。
猜你喜欢
传染病信息(2022年6期)2023-01-12
中国典型病例大全(2022年13期)2022-05-10
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
孩子·小学版(2020年7期)2020-02-24
当代陕西(2019年19期)2019-11-23
少年文艺·开心阅读作文(2018年10期)2018-10-16
小学阅读指南·低年级版(2017年9期)2017-09-12
米娜·女性大世界(2016年10期)2017-02-16
米娜·女性大世界(2016年10期)2017-02-16
