
低氧诱导因子的调控途径和在肠道疾病中的作用*
2019-10-24 11:17滕文彬李玉红祝胜美
中国病理生理杂志 2019年10期
滕文彬, 李玉红, 2△, 祝胜美
(浙江大学绍兴医院 1麻醉科, 2医学研究中心, 浙江 绍兴 312000; 3浙江大学附属第一医院麻醉科, 浙江 杭州 310000)
肠道上皮覆盖着整个胃肠道系统,在成人有大约300平方米的表面积,形成了通往外部的重要屏障,在维持内环境和体内外的平衡起着至关重要的作用。肠道上皮不仅对营养物质的吸收和对非致病性抗原耐受性的形成起着重要作用,同时也受到氧供的调节。近年来,人们越来越认识到组织氧代谢是维持肠道上皮内环境稳态的关键。
正常肠道上皮处于“生理性缺氧”的环境,由于逆流血流和大量细菌的存在,肠道基线动脉氧分压水平低于正常值。肠道独特的生理结构,一侧是高度血管化和氧气充足的黏膜,另一侧是严重缺氧的管腔,因此在上皮层之间有一个陡峭的氧梯度,这种正常肠道上皮独特的耐氧能力,可能是对极低水平氧合状态的一种适应性调节。低氧性应答对机体内的多种生物学进程起着至关重要的作用,是多种疾病如胃肠道疾病,肿瘤和心血管疾病的发病机制。
肠道疾病多发生在肠道“生理性缺氧”的基础上。研究发现,低氧诱导因子(hypoxia-inducible factor,HIF)在维持肠道内环境稳态起着关键作用,对炎症性肠病、肠道肿瘤、缺血再灌注损伤及肠道菌群失调的发展过程中引起的低氧甚至缺氧产生适应性应答,从而维持肠道上皮细胞(intestinal epithelial cells,IECs)的正常功能[1]。
本文将对HIF在IECs中的结构、功能、调节途径及在肠道疾病中作用的相关文献做一综述。
1 HIF的结构、功能和活性调控
HIF作为适应性低氧反应的主要转录调节因子,最初在促红细胞生成素的调节中被发现,后来发现它能转录激活并调节氧平衡和多种与代谢有关的基因[2],包括参与能量代谢、血管生成、细胞凋亡、炎症应答以及其它蛋白质产物增加氧传递或促进对低氧代谢适应的基因,从而成为细胞和系统内对低氧稳态反应的主要调节器。
HIF是碱性螺旋-环-螺旋(basic helix-loop-helix,bHLH)转录因子的Per-ARNT-Sim(PAS)家族成员。HIF的结构呈异源二聚体,由不稳定的α亚基和相对稳定的β亚基组成,每个亚基都包含bHLH-PAS结构域,参与DNA结合的调节[3]。迄今为止,已经发现有3种HIF,即HIF-1、HIF-2和HIF-3。除普遍表达的HIF-1外,哺乳动物中HIF-2也广泛表达,并在红细胞生成、血管化和幼体发育中发挥重要作用[4];HIF-3通过竞争靶基因与低氧反应元件(hypoxic response element,HRE)结合负调控HIF-1和HIF-2[5]。
α亚基是HIF-1的活性亚基,其稳定性和活性均受内环境氧浓度的调控,见图1。在常氧条件下,HIF-1α的转录激活功能被抑制。HIF-1α包含2个高度保守的氧依赖性降解结构域(oxygen-dependent degradation domain,ODD),每个结构域包括脯氨酸残基[6]。脯氨酸羟化酶(proline hydroxylases,PHDs)是Fe2+、α-酮戊二酸(α-ketoglutaric acid,α-KG)和O2依赖的双加氧酶,以O2和α-KG为底物,在特定的脯氨酸残基上(P402和P564)使HIF-1α羟基化,其中一个氧原子与脯氨酰残基结合,另一个与α-KG结合,生成CO2和琥珀酸[6]。羟基化后的脯氨酸被 von Hippel-Lindau (VHL)肿瘤抑制蛋白(pVHL)识别并绑定,然后招募E3泛素连接酶(E3-ubiquitin ligase),并在24S蛋白酶体作用下泛素化靶向降解[7]。
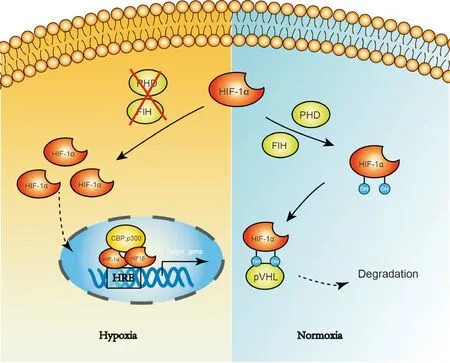
Figure 1. Structure and activity regulation of HIF-1α.
图1 HIF-1α的结构及活性调控
HIF抑制因子1(factor inhibiting HIF-1,FIH-1)是一种天冬酰胺羟化酶,与PHDs类似,也以氧依赖的方式调节HIF的转录活性。FIH-1通过诱导HIF-1α N803位点天冬酰胺残基的羟基化,阻止HIF-1α将转录共激活因子和组蛋白乙酰转移酶p300/CBP招募到HIF-1α的羧基端反式激活结构域(C-terminal transactivation domain,C-TAD)中,阻断与HIF-1α的联系,从而抑制HIF-1α的转录功能[8]。
当机体或细胞受到低氧刺激时,FIH-1和PHDs等氧依赖型酶的活性被抑制,导致胞内HIF-1α累积,然后HIF-1α转位入核与HIF-1β(也称作ARNT)聚合形成异源二聚体,通过招募转录辅活化因子p300/CBP,继而与启动子区域内含有序列5’-A/GCGTG-3’的E-box like HRE结合并调控相关基因的转录表达[7],从而维持氧稳态,使细胞避免低氧损伤或适应低氧环境。
机体或者细胞大多数的低氧应答,都是由HIF介导的。几乎在任何的细胞系中,HIF与至少500个基因位点结合,从而调控相关基因的转录,促进细胞对低氧产生应答[9]。HIF不仅调控多种基因,同时,HIF-1α的基因转录水平、蛋白活性和稳定性也受多种因素调控。除缺氧外,还有其它因素如铁螯合剂、氯化钴、硫化氢和琥珀酸盐等均可影响HIF的活性。
PHDs是铁、α-KG及O2依赖的酶,对HIF-1α修饰后进行蛋白酶体降解。PHDs活性的稳定需要铁,当细胞内铁水平较高时,HIF-1α的活性也被稳定激活[10]。CoCl2的Co2+通过与Fe2+交换来阻止脯氨酸羟化酶和天冬酰胺羟化酶发挥作用,因而作为诱导HIF-1α常用模拟物[10]。H2S通过增强真核生物翻译起始因子2α的磷酸化抑制HIF-1α的转录激活[11]。琥珀酸过量时会因副产物抑制作用而损害PHDs活性,导致HIF-1α的稳定和激活[12],这一作用被α-KG所阻断,α-KG是PHDs的底物,在HIF-1α羟基化作用下生成琥珀酸。
2 HIF的调控途径
2.1核因子κB(nuclear factor-κB,NF-κB)通路 NF-κB是一种快速诱导转录因子,在基因诱导中起着广泛的作用。静息状态下,NF-κB二聚体与NF-κB抑制物(inhibitor of NF-κB,I-κB)家族的蛋白质结合而处于非活性状态[13]。NF-κB活化由I-κB激酶(I-κB kinase,IKK)复合体控制,IKK复合物磷酸化特定丝氨酸残基上的I-κB(主要是I-κBβ)蛋白,诱导其泛素化和蛋白酶体降解,使NF-κB二聚体(p65/p50)在细胞内积聚[13]。随后NF-κB磷酸化的二聚体易位入核,促进HIF-1α的转录[14]。
2.2Janus激酶(Janus kinase,JAK)-信号转导和转录激活子3(signal transducer and activator of transcription 3,STAT3)通路 STAT3是介导细胞因子信号通路的重要转录因子,与多种细胞因子结合并通过JAK/STAT3途径发挥诱导信号转导和基因转录的作用。JAK活化能直接使STAT3磷酸化[15],STAT3转移到细胞核并启动HIF-1α转录[16]。另外,HIF-1α表达上调反过来激活JAK1/2-STAT3信号轴,促进肿瘤干细胞的自我更新[17],表明JAK/STAT3通路与HIF有交互作用。
2.3磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)-蛋白激酶B(protein kinase B,PKB,即AKT)-哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路 PI3K-AKT-mTOR信号通路对HIF-1α有级联放大作用。AKT在下游被PI3K激活,随后磷酸化激活mTOR[18]。磷酸化的mTOR使核糖体蛋白S6激酶(ribosomal protein S6 kinase,S6K,即p70S6K)磷酸化[19],随后磷酸化的p70S6K磷酸化40S核糖体蛋白S6(40S ribosomal protein S6,rpS6)[19],后者对HIF-1α蛋白的合成起到促进作用。mTOR还能使真核细胞翻译起始因子4E结合蛋白1(eukaryotic translation initiation factor 4E-binding protein 1,4E-BP1)磷酸化[19],解除对真核细胞翻译起始因子4E (eukaryotic translation initiation factor 4E,eIF-4E)结合与抑制[20],后者促进HIF-1α蛋白的翻译[21]。
2.4腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)通路 AMPK是一种高度保守的丝氨酸/苏氨酸蛋白激酶复合物,在调节细胞能量平衡中起重要作用。AMPK能抑制mTOR[20],通过p70S6K和4E-BP1对HIF-1α蛋白合成起到抑制作用。AMPK缺失能够提升HIF-1α表达水平,增加糖酵解和合成代谢,增强肿瘤的Warburg效应[22]。
2.5Ras-Raf-MEK-ERK/MAPK通路 Ras-Raf-MEK-ERK/MAPK通路是一种进化保守的通路,参与调控细胞增殖、存活、分化、凋亡和代谢等许多过程[23]。Ras是一种小的GTPase,能被多种受体诱导激活,并与Raf激酶绑定、招募并激活到细胞膜上进行随后的激活;激活后的Raf磷酸化激活MAPK/ERK激酶(MAPK/ERK kinase,MEK),MEK磷酸化激活ERK/MAPK[23];激活后的ERK使4E-BP1、p70S6K和MAPK相互作用激酶(MAPK interacting kinase,MNK)磷酸化。MNK也能直接使eIF-4E磷酸化。最后使mRNA翻译成HIF-1α蛋白加速[8]。
2.6非编码RNA 微小RNAs(microRNAs,miRNAs)是一类小于22个核苷酸的小分子单链非编码RNA,它通过结合3’-非翻译区(3’-untranslated regions,3’-UTR)或氨基酸编码序列来调控。miRNAs介导的转录后调节被认为是最重要的细胞调节方式之一。miR-145通过与p70S6K的3’-UTR结合而抑制p70S6K的转录,从而抑制HIF-1α与血管内皮生长因子(vascular endothelial growth factor,VEGF)的表达[24]。miR-200b通过与HIF-1α的3’-UTR结合抑制其在上皮-间充质转化(epithelial-mesenchymal transition,EMT)中的作用[25]。miR-210通过下调HIF-1α的高度羟基化而降低甘油-3-磷酸脱氢酶1(glycerol-3-phosphate dehydrogenase 1,GPD1L)的水平,从而稳定HIF-1α蛋白[26]。miR-322能通过上调HIF-1α的表达水平促进肿瘤的增殖与迁移[27]。miRNA 497~195簇通过靶向F-box和WD-40结构域蛋白7(F-box and WD-40 domain protein 7,Fbxw7)以及具有跨膜结构域的脯氨酰基4-羟化酶(prolyl 4-hydroxylase, transmembrane,P4HTM),维持内皮Notch活性和HIF-1α的稳定性[28]。
长链非编码RNA(long noncoding RNAs,lncRNAs)是一类长度超过200个核苷酸的非编码RNA。越来越多的证据表明,lncRNAs在基因组印迹、转录激活或失活、RNA剪接、翻译控制和RNA干扰等多个水平上对基因组的调控起着至关重要的作用。LncHIFCAR(long noncoding HIF-1α co-activating RNA)通过直接结合与HIF-1α形成复合物,并促进HIF-1α和p300辅助因子向靶启动子的募集,来共同激活HIF-1α[29]。 lncRNA CPS1-IT1(CPS1 intronic transcript 1)通过与热休克蛋白90(heat shock protein 90,HSP90)竞争性结合抑制HIF-1α的表达[30],从而抑制肿瘤的转移和肠上皮细胞的转分化[31]。lncRNA-NUTF2P3-001 可以调节HIF与HRE的上游启动原件 RAS,并在肿瘤中呈高表达[32]。
由此可见,HIF受到多种多层次、多位点、多过程调控,为多种疾病治疗提供很多潜在靶点。
3 HIF与相关疾病
HIF在肠道中持续表达,是调节肠道代谢的重要转录因子,肠道多种疾病的发生常常伴随HIF活性增强或降低,影响肠道机械屏障功能、生物屏障以及黏液屏障等,从而出现各种病理或者病理生理改变。
3.1炎症性肠病(inflammatory bowel disease,IBD) IBD如溃疡性结肠炎(ulcerative colitis,UC)和克罗恩病(Crohn disease,CD)等的患者对肠腔内微生物的炎症反应加剧,肠道黏膜产生大量炎症细胞因子和炎症介质,IECs被破坏、甚至发生凋亡,致使肠黏膜屏障功能障碍,加重病情。临床研究显示,在UC或者CD患者的IECs中,黏膜缺氧促进HIF-1α的高表达[33]。HIF-1α激活能促进炎症消退,还能通过促进肠黏膜屏障的修复,防止上皮细胞凋亡,从而可能减轻病情。
HIF-1α的稳定在肠道中具有保护性作用。在小鼠结肠炎模型中,肠道上皮HIF-1α突变的小鼠,HIF-1α条件性功能丧失,IECs的条件性损伤加剧,结肠炎的症状加重,比如死亡率升高,体重减轻,结肠长度缩短;而肠道上皮pVHL突变的小鼠,DNA碱基水平发生改变,pVHL条件性功能失活,HIF-1α持续激活,肠道屏障保护基因(比如多药耐药基因1α、肠三叶因子、CD73等)的表达水平增加,体内结肠炎期间肠道屏障功能的损伤减轻[34]。另一研究中,HIF-1α缺失的小鼠体重下降明显,肠道炎症程度加重,促炎细胞因子水平升高,黏蛋白增加[35]。而通过抑制HIF-1α的降解,增加HIF-1α的稳定性能显著降低炎症所致的IECs损伤程度[36]。这些研究表明,HIF-1α是肠道屏障的保护性因素。因此,HIF-1α是一种很有希望作为治疗药物靶点的候选分子。
3.2肠道肿瘤 缺氧是几乎所有实体肿瘤微环境中的一种主要现象,因为肿瘤细胞的膨胀速度迅速地超过滋养血管的发生,再加之肿瘤血管的功能紊乱,进一步加重缺氧。研究表明,HIF-1α在肠道肿瘤中高表达,与肿瘤的发生、浸润、转移、化疗耐药和辐射抗性高度相关[37]。HIF-1α高表达是结肠癌总体生存率和无进展生存率的独立影响因素,抑制HIF-1α表达可显著抑制肿瘤生长[38]。研究显示,HIF-1α在肿瘤血管生成中起着重要作用。缺氧条件下,HIF-1α是VEGF表达的关键调节因子,HIF-1α通过与HREs结合激活VEGF的转录[39]。直结肠癌进展中,肿瘤出牙与直结肠癌形成前的低氧诱导的小血管形成有关,出芽细胞表达HIF-1α介导缺氧性肿瘤表型[40]。在裸鼠中,过表达PHD1 可明显抑制肿瘤生长,肿瘤生长受抑与坏死增加和微血管密度显著降低有关[41]。
EMT是上皮细胞转化为具有间充质表型细胞的过程,是癌症转移的关键步骤之一。HIF-1α在EMT中同样发挥重要作用。在体或离体实验中,通过抑制HIF-1α诱导的自噬作用抑制大肠癌EMT和转移[31]。
3.3缺血再灌注(ischaemia-reperfusion,I/R)损伤 I/R是引起肠道损伤常见原因,可导致组织损伤、炎症反应以及IECs凋亡。肠道I/R常见病因有绞窄性疝、肠系膜动脉阻塞或循环衰竭等。研究表明,在肠道I/R中,HIF-1α主要通过腺苷调节肠道的保护功能。低氧环境下HIF-1α能诱导细胞外腺苷生成,增加信号传导,具体是通过A2B腺苷受体,发挥对肠道的保护作用[42]。
肠系膜上动脉闭塞(superior mesenteric artery occlusion,SMAO)60 min的小鼠模型中,条件性敲除肠道HIF-1α会加重I/R诱导的损伤;应用PHDs抑制剂DMOG能减轻I/R对肠道的损伤作用[42]。在HIF-1α条件性缺失的小鼠和体外条件性敲除HIF-1α的IECs中,缺氧对平衡性核苷转运体的抑制作用消失,表明HIF-1α还能通过抑制核苷转运体抑制腺苷再摄取和通过腺苷激酶抑制腺苷代谢以增加细胞外腺苷浓度[43]。
Kannan等[44]研究在SMAO的肠I/R损伤模型(45 min SMAO再灌注3 h)中,使用部分HIF-1α缺陷的小鼠,结果发现HIF-1α激活与肠道I/R损伤直接相关;HIF-1α部分缺陷通过降低SMAO诱导的肠通透性增加、脂质过氧化、黏膜caspase-3活性及IL-1β水平的升高,减轻肠I/R损伤的作用,表明I/R损伤的持续时间和严重程度决定HIF-1α是否具有肠道保护或有害作用。因此,HIF-1α是肠道I/R的保护因子,有可能成为治疗I/R损伤的潜在靶点。
3.4肠道细菌与感染 肠道内多种微生物共生,形成一个动态的微生物环境。IECs对病原菌的入侵具有高度的警惕性,是抵抗病原体入侵和感染的重要屏障。当大量的外源性致病菌入侵肠道或者内源性菌群失调,会进一步损伤IECs,甚至诱导发生凋亡,成为肠道疾病的来源。发生感染时,细胞的氧需大于氧供是低氧应激反应的一种常见现象。低氧条件下,细菌大量摄取铁,抑制PHDs活性,使HIF-1α稳定,诱导HIF靶基因的表达[45]。另一研究揭示,在一些肠道菌属(如沙门氏菌、耶尔森菌、肠杆菌)中的铁载体也具有稳定HIF-1α的作用[46]。
肠道HIF-1α可通过诱导多种肠道保护因子抵抗病原菌的侵袭作用。髓系细胞系中条件性缺失HIF-1α小鼠的杀菌活性降低,容易导致感染在全身扩散;相反,通过敲除pVHL或激活HIF-1α能使髓系细胞产生防御因子,提高杀菌能力[47]。肠道上皮细胞缺失HIF-1α的小鼠对口服小肠结肠炎耶尔森菌的易感性增高,细菌介导的HIF-1α激活可能代表宿主启动对病原菌的防御机制[46]。在离体Caco-2细胞模型中,HIF-1α稳定剂DMOG通过减少宿主β-1整合素在细胞表面的定位,减少小肠结肠炎耶尔森菌对肠上皮细胞的内化而降低其侵袭力[48]。
肠道微生物能产生多种代谢产物,尤其是短链脂肪酸,包括丁酸盐、丙酸盐和醋酸盐,对肠道起保护作用。丁酸盐是细菌首要代谢产物,在正常的肠道组织中,高达30%的能量可能来自丁酸盐的代谢,并在结肠黏膜中起到多种功能。丁酸盐能刺激IECs代谢,增加O2的消耗,使HIF-1α保持稳定,增强IECs功能[49]。通过使用抗生素,减少微生物,能降低结肠的丁酸水平和HIF-1α的表达,而通过补充丁酸能恢复恢复HIF-1α的表达水平[50]。此外,在条件性缺失HIF的细胞中,丁酸失去了对屏障的保护作用,表明丁酸对肠黏膜屏障的保护功能可能通过HIF起作用[49]。这表明短链脂肪酸通过HIF-1α信号轴促进肠道的保护功能。
4 总结与展望
近年来,分析工具和基因组技术发展的进步实现了对缺氧和缺氧诱导转录因子的生物学研究,提高了人们对生理和病理状态中氧稳态的认知。HIF在组织氧代谢中的作用一直是许多学者关注的焦点。HIF对IECs代谢的作用越来越受到重视,其对维持肠黏膜细胞的正常功能、肠道屏障的完整性及肠道微环境的稳定性具有重要意义。对HIF结构、作用机制及其影响因素的深入研究为肠道炎症、感染、肿瘤、缺血再灌注等发病机制提供新的思路,并通过对HIF调节机制的进一步研究探索上述疾病新的治疗靶点。
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2022年8期)2022-12-07
江苏安全生产(2022年8期)2022-11-01
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
小资CHIC!ELEGANCE(2021年36期)2021-10-15
天津医科大学学报(2021年3期)2021-07-21
艺术评鉴(2020年5期)2020-04-30
中华老年口腔医学杂志(2019年2期)2019-04-28
人大建设(2018年10期)2018-12-07
分析化学(2017年12期)2017-12-25
