
肺炎克雷伯杆菌表达吡咯喹啉醌依赖性聚乙烯醇脱氢酶
2019-10-16 09:01:06弥志伟孙忠义田平芳
北京化工大学学报(自然科学版) 2019年5期
弥志伟 孙忠义 赵 鹏 田平芳
(北京化工大学 生命科学与技术学院, 北京 100029)
引 言
聚乙烯醇(PVA)广泛用于纺织、造纸和涂料等行业,纺织工业产生的退浆废水残留大量PVA,造成严重的环境污染。相比于高温碱法,采用PVA生物降解法进行退浆,一方面有利于减少废物排放,另一方面有利于保护织物组织[1-2]。目前报道的PVA生物降解方式主要有两种:一种是PVA氧化酶(SAO)与氧化型PVA水解酶(OPH)组合降解,另一种是PVA脱氢酶(PVADH)与OPH组合降解[3-4]。
目前,已有Pseudomonassp. VM15C和Sphingopyxissp. 113P3两个菌株的pvadh基因得到确认[5-6],后者在大肠杆菌和毕赤酵母中表达后酶活分别达到194 U/mL[7]和8 464 U/mL[8],但是在大肠杆菌中易形成包涵体,需进行复杂的包涵体溶解和蛋白复性,而近一半蛋白无法恢复活性[7];在毕赤酵母中则会因为体内蛋白酶的作用发生血红素结构域的切割,无法得到完整的PVADH[8]。成熟的PVADH位于周质空间,属于吡咯喹啉醌(pyrroloquinoline quinone,PQQ)依赖性醌酶,而大肠杆菌和毕赤酵母中生产的PVADH不含辅酶,需要结合其他菌株或人为提供的PQQ才具有催化活性。由于PQQ价格昂贵,增加了PVADH使用的成本,限制了其工业化应用。
作为一种热点工业微生物[9],肺炎克雷伯杆菌(Klebsiellapneumoniae)具有如下优势:(1)含有pqqABCDEF基因簇(简称pqq),能够天然生产辅酶PQQ[10],节省外源添加PQQ的成本[11];(2)作为原核宿主菌,在生产过程中发生蛋白酶切割PVADH的可能性小;(3)发酵成本低,生长快,能高效利用甘油生产1, 3-丙二醇、3-羟基丙酸等大宗化学品[12-13],附加产值高。本文拟采用K.pneumoniae作为PVADH的生产菌株,实现PQQ- PVADH全酶的高效生物合成。
1 实验部分
1.1 材料
1.1.1菌株与载体
K.pneumoniaeDSM2026,大肠杆菌E.coliDH5α,表达载体pET- 3tac(将pET- 28a的T7启动子替换为3个串联的tac启动子),pET- 15A- cm(将pET- 28a的复制子替换为15A,抗性基因替换为氯霉素(cm)抗性基因),均为本实验室保存。
1.1.2培养基
LB培养基:蛋白胨10 g/L;酵母粉5 g/L;NaCl 10 g/L;pH 7.0。固体培养基添加15 g/L琼脂。
甘油发酵培养基:甘油30 g/L,酵母粉3 g/L,KH2PO41.3 g/L,K2HPO4·3H2O 3.4 g/L,(NH4)2SO44.0 g/L,MgSO4·7H2O 0.5 g/L,CaCO30.1 g/L,微量元素1.25 mL/L[14]。
抗性培养基中加入50 μg/mL硫酸卡那霉素(kan)或17 μg/mL cm。
1.1.3主要试剂
限制性内切酶和DNA连接酶购买自NEB(北京)有限公司;Taq DNA聚合酶、质粒提取试剂盒、PCR产物回收试剂盒和DNA marker购买自北京博迈德基因技术有限公司;BCA蛋白定量试剂盒购买自北京全式金生物技术有限公司;引物和优化后的pvadh由华大基因(北京)合成。DNA测序由北京睿博兴科生物技术有限公司完成。其余常规化学试剂均为国产分析纯。
1.2 方法
1.2.1重组菌构建
以National Center for Biotechnology Information(NCBI)报道的Sphingopyxissp. 113P3pvadh基因序列(GenBank accession No. JQ235753)为模板,使用DNAMAN软件按照K.pneumoniae密码子偏好性优化,并添加EcoR I和XhoI两个酶切位点。将优化后的pvadh与pET- 3tac质粒双酶切,经T4连接酶催化连接后,得到重组质粒pET- 3tac-pvadh。
待重组质粒转化感受态E.coliDH5α经抗性筛选、菌落PCR验证和测序确认后,将正确的质粒电转至感受态K.pneumoniaeDSM2026,得到重组菌Kp(pET- 3tac-pvadh)。
为了解决超表达PVADH后PQQ供应不足的问题,构建另一个超表达PQQ的载体。根据GenBank报道的pqq基因序列设计引物,两端引入EcoR I和Hind III 酶切位点(表1),克隆K.pneumoniae的pqq基因簇,PCR反应参数:94 ℃,5 min;94 ℃,40 s;55 ℃,40 s;72 ℃,6 min,30个循环;72 ℃,10 min,16 ℃保存。将PCR产物和质粒pET- 15A-cm通过EcoR I和Hind III双酶切,经T4连接酶催化连接后,得到重组质粒pET- 15A-cm-pqq,将其转化至E.coliDH5α感受态,抗性筛选得到的单菌落采用pET载体通用上游引物pET- up和pqqB基因下游引物pqqB- R进行菌落PCR验证,经测序确认后,将pET- 15A-cm-pqq电转至Kp(pET- 3tac-pvadh)中,得到同时超表达pvadh和pqq的双质粒菌株Kp(pvadh+pqq),在含50 μg/mL kan和17 μg/mL cm的LB双抗固体培养基上培养,通过菌落PCR筛选同时含有两个质粒的单菌落。
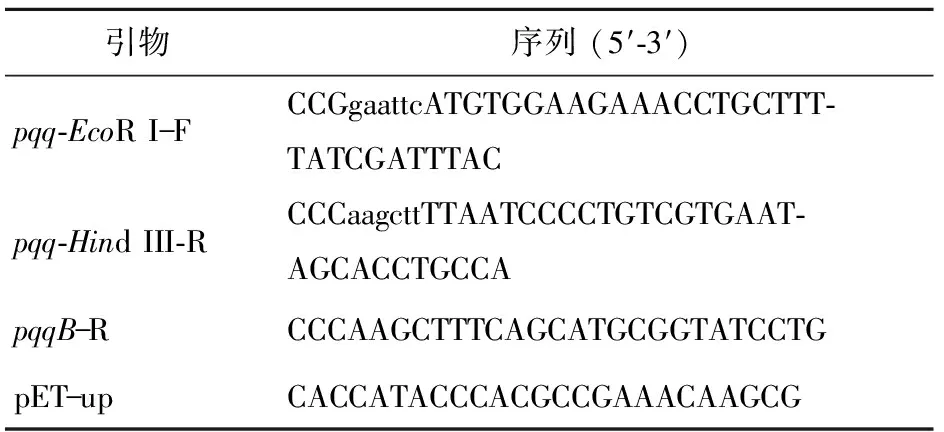
表1 引物序列
小写字母表示酶切位点;F为上游引物,R为下游引物。
1.2.2摇瓶发酵及优化
菌株活化培养 在含4 mL LB培养基的试管中,按1%接种量(体积分数)加入保存的菌液,37 ℃,200 r/min,恒温震荡培养12 h。
摇瓶发酵培养 在含100 mL发酵培养基的250 mL三角摇瓶中,按1%接种量(体积分数)加入活化后菌液,37 ℃,200 r/min,微氧发酵。接种2 h后,加入0.5 mmol/L(异丙基硫代半乳糖苷)到发酵液中。
数据采集 每3 h取样,测定菌液在600 nm波长处的吸光值A600,24 h后取菌液制备粗酶液,测PVADH酶活。
发酵优化 通过单因素试验依次对甘油添加量、IPTG添加量、IPTG添加时间、发酵温度进行优化。
1.2.3PVADH粗酶液制备和优化
PVADH前体含由25个氨基酸组成的信号肽,在帮助PVADH跨膜之后被切除。成熟的PVADH属于周质蛋白,获得周质蛋白的方法有超声破碎法和渗透休克法[15]。本文对比了两种方法的效果。
(1)超声破碎法获得PVADH粗酶液摇瓶发酵24 h后,取500 mL发酵液于4 ℃ 12 000 r/min下离心10 min,收集菌体,用PBS缓冲液洗涤1次后重悬于含1 mmol/L蛋白酶抑制剂苯甲基磺酰氟(PMSF)的50 mL PBS缓冲液中。取25 mL重悬液置于50 mL离心管中,冰浴30 min,在冰水保护下进行间歇式超声处理,超声15 s,暂停45 s,反复10次左右,直至菌液变为澄清均质溶液。
(2)渗透休克法获得PVADH粗酶液500 mL发酵液,离心去上清液,菌体用PBS缓冲液洗涤1次后重悬于50 mL 30%蔗糖溶液中,室温放置10 min,4 ℃离心收集菌体,再重悬于50 mL预冷的5 mmol/L MgSO4溶液中(含1 mmol/L PMSF),吸打混匀后置于冰上20 min,4 ℃离心,得上清液,即周质蛋白溶液。
1.2.4PVADH酶活性检测
参考文献[16]中的方法对PVADH的酶活性进行检测,PVADH酶活定义为酶活检测体系中,每min减少1 nmoL 2, 6-二氯酚靛酚钠(DCIP)(ε=1.91×104mmol/(L·cm))所需要的酶量为一个活力单位,单位U/mL。
2 结果与讨论
2.1 pvadh密码子的优化及其在K. pneumoniae中的异源表达
不同物种对同义密码子的使用具有偏好,利用这一特点,有针对性地进行密码子优化,可帮助外源蛋白高效表达。为此,将Sphingopyxissp. 113P3的pvadh序列根据K.pneumoniae的密码子偏好性进行优化,优化后的pvadh密码子适应指数(CAI)从0.7提升到0.95,理论上有利于提高蛋白的合成速率和数量。
提取pET- 3tac质粒(图1(a)),与合成的pvadh经EcoR I和XhoI双酶切并连接后,转入E.coliDH5α感受态。菌落PCR采用pET载体的通用引物pET- up与pvadh的下游引物pvadh-XhoI- R,图1(b)中3号菌株为阳性重组菌。经测序确认后,将质粒pET- 3tac-pvadh电转至K.pneumoniae感受态中,经菌落PCR确认(图1(c)),即得到重组菌Kp(pET- 3tac-pvadh)。
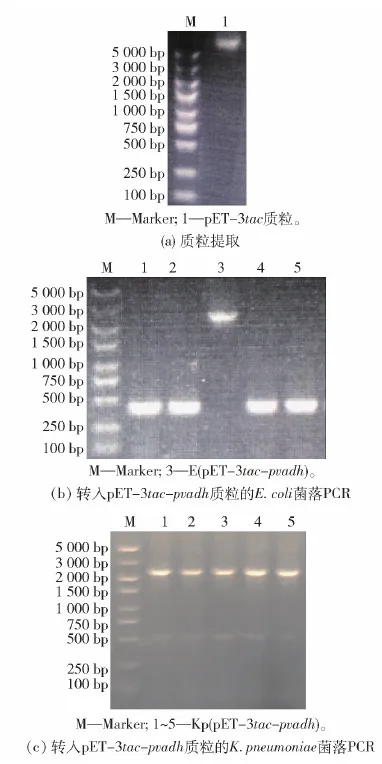
图1 重组菌株Kp(pET- 3tac- pvadh)的构建Fig.1 Construction of the recombinant strain Kp(pET- 3tac- pvadh)
发酵培养Kp(pET- 3tac-pvadh),15 h取样作SDS- PAGE蛋白电泳。如图2所示,重组菌在67 kDa的位置出现特异性条带,符合切除信号肽之后PVADH蛋白的大小,说明实验成功实现了PVADH在K.pneumoniae中的异源表达。
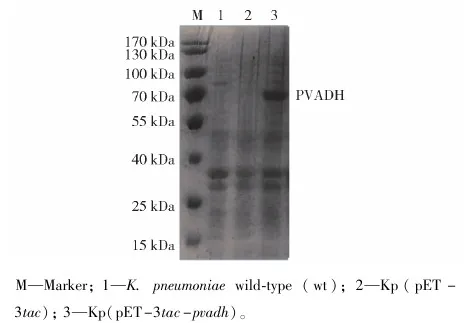
M—Marker; 1—K. pneumoniae wild-type (wt); 2—Kp(pET- 3tac); 3—Kp(pET- 3tac- pvadh)。图2 超表达pavdh肺炎克雷伯重组菌的SDS- PAGE蛋白电泳结果Fig.2 SDS- PAGE analysis of recombinant K. pneumoniae strain overexpressing pavdh
2.2 PVADH粗酶液制备及酶活测定结果
将渗透休克法和超声破碎法分离PVADH蛋白的效果进行比较(表2),可以看出,渗透休克法获得粗酶液的总蛋白和酶活都不如超声法,但是因为渗透休克法获得的上清液中主要是周质蛋白,PVADH比活是超声破碎法得到的粗酶液的3.7倍,所以后
表2 渗透休克法和超声破碎法获得PVADH粗酶液的效果比较续选择渗透休克法制备PVADH粗酶液。
Table 2 Characteristics of the PVADH crude enzyme obtained by osmotic shock and ultrasonic fragmentation
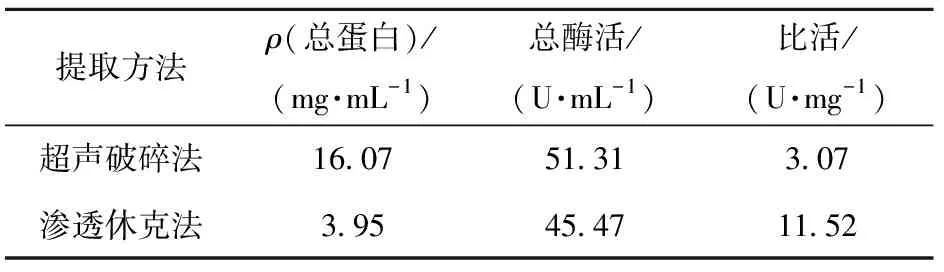
提取方法ρ(总蛋白)/(mg·mL-1)总酶活/(U·mL-1)比活/(U·mg-1)超声破碎法16.0751.313.07渗透休克法3.9545.4711.52
2.3 辅酶PQQ的再生
摇瓶发酵Kp(pET- 3tac-pvadh),24 h的PVADH酶活为45.47 U/mL(图3)。前人在K.pneumoniae中超表达可溶性葡萄糖脱氢酶(sGDH)后发现辅酶PQQ不能满足sGDH的需求,额外添加PQQ能明显提升酶活[12]。
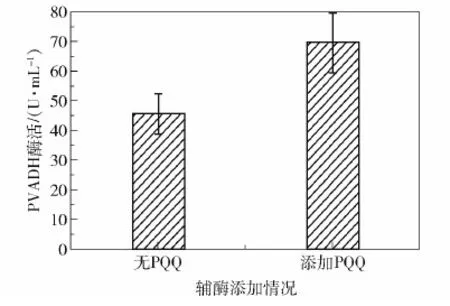
图3 添加或不添加PQQ时PVADH的酶活Fig.3 PVADH activity with or without PQQ addition
本文实验也发现,添加6 μmol/L的PQQ有助于提高Kp(pET- 3tac-pvadh)酶活,相比不添加PQQ的粗酶液,添加PQQ的粗酶液酶活提高了55.6%(69.6 U/mL),这说明超表达pvadh之后,辅酶PQQ的供给相对不足,所以考虑通过辅酶再生提高Kp(pET- 3tac-pvadh)中PQQ产量。为了避免相同复制子在同一宿主中发生质粒不相容问题,采用了本实验室构建的pET- 15A-cm载体超表达pqq。提取K.pneumoniae基因组(图4(a)),克隆pqq(图4(b)),通过EcoR I和Hind III位点插入pET- 15A-cm,获得pET- 15A-cm-pqq质粒,将其电转入Kp(pET- 3tac-pvadh)感受态中,通过两组菌落PCR分别验证两个质粒的存在:①采用通用上游引物 pET- up和pqqB- R验证pET- 15A-cm-pqq质粒,正确条带大小为1 500 bp(图4(c));②采用pET- up与pvadh- xhoI- R验证pET- 3tac-pvadh质粒,正确条带大小为2 300 bp(图4(d))。包含两个质粒的菌株即为Kp(pvadh+pqq)。
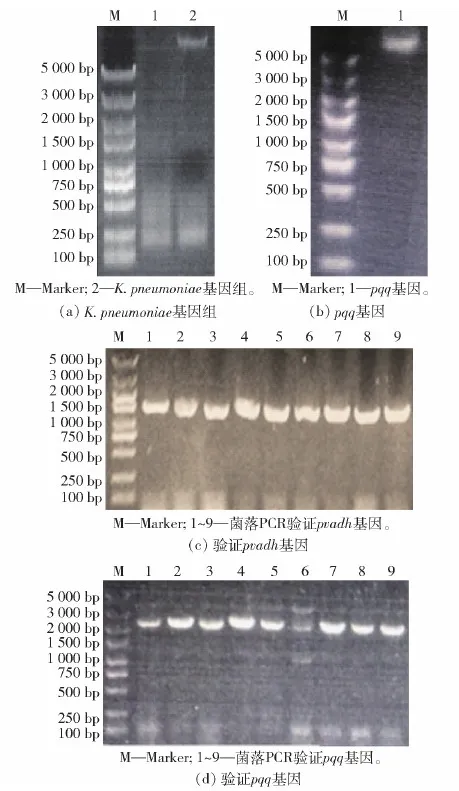
图4 同时超表达pvadh和pqq的K. pneumoniae重组菌构建Fig.4 Construction of recombinant K. pneumoniae strain coexpressing genes pvadh and pqq
在摇瓶发酵培养阶段,与野生型K.pneumoniae及Kp(pET- 3tac-pvadh)相比,Kp(pvadh+pqq)在开始时显示出明显的生长负荷,但随着时间的延长其生长速度加快,9 h后生物量接近野生型和Kp(pET- 3tac-pvadh)(图5(a))。24 h时检测酶活,发现Kp(pvadh+pqq)的PVADH酶活明显提高,在没有添加PQQ的情况下酶活达到了67.5 U/mL(图5(b))。以上结果说明辅酶再生很好地满足了PVADH对PQQ的需求。
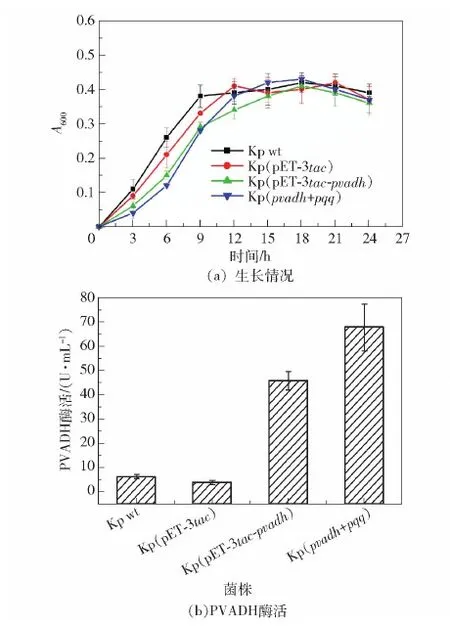
图5 Kp(pET- 3tac- pvadh)和Kp(pvadh+pqq)的生长情况与PVADH酶活Fig.5 Growth and PVADH activities of Kp(pvadh+pqq) and Kp(pET- 3tac- pvadh)
2.4 发酵优化提高酶活
为进一步提高K.pneumoniae中PQQ- PVADH产量,对Kp(pvadh+pqq)的发酵体系进行单因素优化,具体参数包括甘油浓度、IPTG添加量和添加时间及温度。
首先是甘油浓度的优化。K.pneumoniae能高效代谢甘油[13],因此选择甘油作为Kp(pvadh+pqq)的碳源。实验对比了不同甘油浓度对菌体生长和PVADH酶活的影响,结果如图6所示,发现随着甘油质量浓度从10 g/L升至40 g/L,Kp(pvadh+pqq)的生长量持续攀升,PVADH酶活也随着生物量一同升高;但当甘油质量浓度超过40 g/L后,Kp(pvadh+pqq)的生长量和PVADH酶活不升反降,推测可能是高浓度甘油增加了培养基黏度,影响了菌体生长。
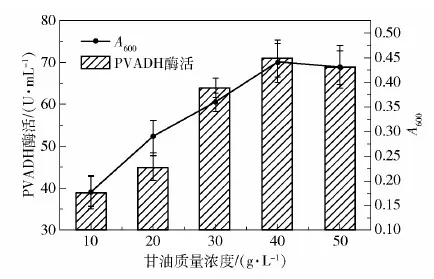
图6 Kp(pvadh+pqq)发酵中甘油浓度的优化Fig.6 Optimization of glycerol concentration in fermentation of Kp(pvadh+pqq)
然后是IPTG添加量的优化。构建的两个质粒分别超表达pvadh和pqq,都需要IPTG诱导表达,适度增加IPTG浓度有助于提升两个基因的表达量。从图7可知,随着IPTG添加量的升高,菌体生物量持续下降,表明IPTG对菌体生长有明显抑制作用;但PVADH的酶活却有明显提升,说明IPTG诱导对pvadh和pqq的高效表达至关重要,结果显示加入0.6 mmol/L IPTG时PVADH酶活最高。
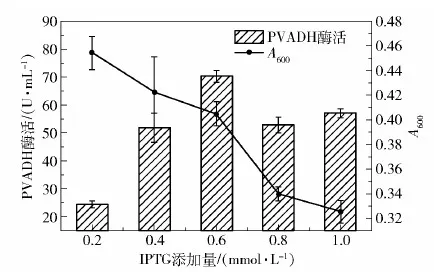
图7 Kp(pvadh+pqq)发酵中IPTG添加量的优化Fig.7 Optimization of IPTG addition in Kp(pvadh+pqq) fermentation
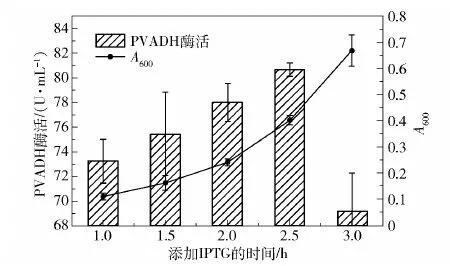
图8 Kp(pvadh+pqq)发酵中IPTG添加时间的优化Fig.8 Optimization of time to add IPTG in Kp(pvadh+pqq) fermentation
之后是诱导剂添加时间的优化。如图8所示,接种后1 h到3 h,Kp(pvadh+pqq)的生物量A600从0.11缓慢提升到0.67,在A600=0.1~0.4区间内,生物量越大,添加IPTG对24 h后PVADH酶活的提升效果越明显,但当A600达到0.67时添加IPTG,PVADH酶活明显降低,所以选择发酵后2.5 h(A600=0.4)添加IPTG。
最后是发酵温度的优化。如图9所示,随着温度从25 ℃升高到37 ℃,Kp(pvadh+pqq)的生物量和PVADH酶活均有明显提高;但当温度升至42 ℃后,菌体生长严重受阻,生长量和PVADH酶活骤降,所以选择37 ℃作为Kp(pvadh+pqq)的发酵温度。
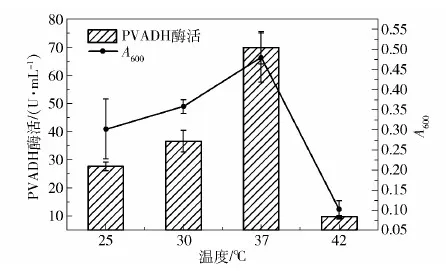
图9 Kp(pvadh+pqq)发酵温度的优化Fig.9 Optimization of temperature in Kp(pvadh+pqq) fermentation
综合以上结果,优化后的条件为:40 g/L甘油,37 ℃,2.5 h后添加0.6 mmol/L IPTG进行发酵,24 h时PVADH酶活可达到80.7 U/mL。
3 结论
成功在PQQ天然生产菌K.pneumoniae中异源表达了pvadh,通过密码子优化实现了PVADH的高效表达,PVADH酶活达到45.47 U/mL;为解决PQQ供应不足的问题,构建双质粒表达系统同时超表达pqq基因簇,提高宿主菌的PQQ合成量,进一步补充了辅酶PQQ,PVADH酶活达到67.5 U/mL;最后通过发酵条件优化,PVADH酶活达到80.7 U/mL。
目前K.pneumoniae尚未通过基因工程优化其基因组,不具备商业化底盘微生物如E.coli和P.pastoris在基因表达方面的优势,因此其PVADH产量相对较低。但作为一种PQQ天然生产菌,K.pneumoniae在生产PVADH的同时节省了添加辅酶PQQ的高昂成本,为PQQ- PVADH的生产提供了另一个选择方案。随着K.pneumoniae表达体系的不断优化,PQQ- PVADH产量还将进一步提升。
猜你喜欢
中老年保健(2022年4期)2022-08-22 02:58:30
生物学通报(2020年11期)2020-10-22 01:20:20
中成药(2018年7期)2018-08-04 06:04:10
能源(2017年7期)2018-01-19 05:05:04
中学科技(2017年11期)2017-12-26 10:14:01
中国民族医药杂志(2016年4期)2016-05-09 07:41:11
中外医疗(2015年11期)2016-01-04 03:58:49
生命科学研究(2014年1期)2014-04-29 00:44:03
西部中医药(2014年6期)2014-03-11 16:07:47
中国中医药现代远程教育(2014年23期)2014-03-01 04:33:21
