
Dynamic changes of root proteome reveal diverse responsive proteins in maize subjected to cadmium stress
2019-10-10 06:08RENWenLIUYaZHOUMiaoyiSHIZiWANGTianyuZHAOJiuranLIYu
Journal of Integrative Agriculture 2019年10期
REN Wen , LIU Ya, ZHOU Miao-yi, SHI Zi, WANG Tian-yu ZHAO Jiu-ran, LI Yu
1 Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, P.R.China
2 Maize Research Center, Beijing Academy of Agriculture & Forestry Sciences/Beijing Key Laboratory of Maize DNA Fingerprinting and Molecular Breeding, Beijing 100097, P.R.China
Abstract Toxic symptoms and tolerance mechanisms of heavy metal in maize are well documented. However, limited information is available regarding the changes in the proteome of maize seedling roots in response to cadmium (Cd) stress. Here, we employed an iTRAQ-based quantitative proteomic approach to characterize the dynamic alterations in the root proteome during early developmental in maize seedling. We conducted our proteomic experiments in three-day seedling subjected to Cd stress, using roots in four time points. We identified a total of 733, 307, 499, and 576 differentially abundant proteins after 12, 24, 48, or 72 h of treatment, respectively. These proteins displayed different functions, such as ribosomal synthesis,reactive oxygen species homeostasis, cell wall organization, cellular metabolism, and carbohydrate and energy metabolism.Of the 166 and 177 proteins with higher and lower abundance identified in at least two time points, 14 were common for three time points. We selected nine proteins to verify their expression using quantitative real-time PCR. Proteins involved in the ribosome pathway were especially responsive to Cd stress. Functional characterization of the proteins and the pathways identified in this study could help our understanding of the complicated molecular mechanism involved in Cd stress responses and create a list of candidate gene responsible for Cd tolerance in maize seeding roots.
Keywords: cadmium stress, iTRAQ proteomics, maize seedling roots
1. Introduction
Heavy metals in plants represent either as essential micronutrients or act as toxic compounds (Polle and Schützendübel 2003). Cadmium (Cd) is one of the most highly toxic environmental pollutants. Excessive Cd causes serious problems to all organisms (Benavides et al. 2005).Plants absorb Cd via the roots, transport it to the shoots via the xylem, and accumulate it in seeds (Yang et al. 2016).In high concentration conditions, Cd causes a drop in photosynthetic capacity, water loss that results in a reduction of the length of roots and shoots, leaf bleaching (Rascio et al.1993), necrosis, and wilting (Wang et al. 2007).
In order to alleviate stress damages and survive in heavy metal-contaminated soil, plants have developed various of defense strategies to regulate the uptake, mobilization, and intracellular concentration of toxic heavy metal ions (Hossain and Komatsu 2012). Tremendous efforts have been made during last decades to understand the mechanism behind toxicity responses and detoxification during exposure to Cd. To identify heavy metal-responsive genes or proteins is a fundamental step in characterizing the mechanisms of heavy metal toxicity responses and detoxification (Ahsan et al. 2009). In recent years, with the development of high throughput sequencing, previous studies employed transcriptomic analysis to investigate Cd-responsive genes in different species (Peng et al. 2015; Xu et al. 2015; Liu et al. 2017), identifying a large number of differentially expressed genes (DEGs) responding to Cd treatment.DEGs participating in several processes such as cell wall biosynthesis, glutathione (GSH) metabolism, tricarboxylic acid (TCA) cycle, and antioxidant system that probably play critical roles in cell wall binding, vacuole sequestration,and detoxification, identified in Cosmos bipinnatus Cav. by transcriptome analysis (Liu et al. 2017). In Sorghum bicolor(L.) Moench, Feng et al. (2018) showed many Cd-responsive DEGs are involved in cell wall modification, heavy metal transport, and phenylpropanoid biosynthesis. Isolation of transcripts from radish (Raphanus sativus L.) root under Cd stress indicates that DEGs are predominately involved in glucosinolate biosynthesis as well as cysteine and methionine-related pathways (Xu et al. 2015). Moreover,de novo sequencing of root transcriptome revealed a schematic model of DEGs and microRNAs-involved in Cd-responsive regulatory network (Xu et al. 2015). Using comparative an RNAseq-based approach, Peng et al.(2015) investigated transcriptomic changes during maize roots development under Cd pollution, and identified many DEGs encoding stress and defense responses proteins,transporters, as well as transcription factors.
Transcriptomics provide an indispensable approach for unraveling gene expression networks, as mRNA expression levels reflects the function of corresponding proteins.However, mRNA levels do not fully represent protein levels,because of the activity of the post transcription machinery or protein degradation (Haider and Pal 2013). Proteomic technique, which offers a link between gene expression and cell metabolism, is a powerful tool for the analysis of molecular mechanism of plant response against biotic and abotic stresses (Wang Y et al. 2016). Isobaric tags for relative and absolute quantitation (iTRAQ), a proteomic approach, provides reliable quantitative measurements of protein quantities (Karp et al. 2010). This method significantly improves proteomic analysis in combination with Gene Ontology (GO) and pathway studies (Xu et al. 2016).iTRAQ studies identify many differentially abundant proteins(DAPs) after exposure to heavy metals such as manganese(Mn, You et al. 2014), lead (Pb, Li et al. 2015; Wang Y et al.2016), copper (Cu), and Cd (Mota et al. 2015). In maize,iTRAQ-based approaches found numerous DAPs involved in various pathways under the stress such as chilling(Wang X et al. 2016), Pb (Li et al. 2015), osmotic (Hu et al.2015a), waterlogging (Yu et al. 2014), drought (Benešová et al. 2012), or heat (Hu et al. 2015b). However, no data is present regarding protein changes in response to Cd stress in maize seedling roots in early developing stages.
Here, we employed an iTRAQ-based proteomics approach to identify protein changes in maize root at 12,24, 48, or 72 h upon exposure to Cd. The candidate genes identified in this study could improve our understanding of the regulatory networks in the response to Cd stress in maize, and provide useful insights for biotechnological applications to increase plant fitness in field sites with heavy metal pollution.
2. Materials and methods
2.1. Plant materials and cadmium treatment
The inbred line Jing724 is the maternal parent of Jingke 968, which is one of the elite maize varieties in China(Su et al. 2016). To investigate the mechanism of Cd stress will provide useful information to breed new varieties under Jing724 background that adapts to various environments.Healthy and equal-sized seeds of the maize inbred line Jing724 were sterilized using 10% H2O2for 1 h, then soaked in distilled water for 24 h, and germinated in the dark for 3 d at 25°C. Seedlings were grown in modified Hoagland’s solution (Pál et al. 2005), with a 14-h/10-h light/dark cycle at 24-26°C. After 3 d of cultivation, seedlings were separated randomly into control and treatment groups. The treatment group seedlings were treated with 200 mg L-1CdCl2·2.5H2O solution, and the control group seedlings were grown in Cd-free Hoagland’s solution. The roots of seedlings in both groups were harvested separately at 12, 24, 48, or 72 h after Cd treatment. Treatments were performed in two independent biological replicates (R1 and R2). To investigate the enrichment capacity of Cd in Jing724, we used B73 (the reference variety of maize) as control. Cd treatment for B73 was same as for Jing724.
2.2. Maize plant characteristics and root Cd content
We selected samples at 48 h as representations to show Cd uptake and accumulation characteristics. Root tissues of both Jing724 and B73 were oven-dried at 80°C, ashed at 550°C, then 0.0500 g of root tissue (dry weight) was dissolved in 6 mL concentrated HNO3and 2 mL 30% H2O2,then brought to 50 mL adding ddH2O, before analyzing the Cd content by inductively coupled plasma mass spectrometry (ICP-MS) (Varian, Torino, Italy).
2.3. Protein preparation and digestion
For each sample, approximately 0.5 g of fresh root tissue of Jing724 was pulverized in liquid nitrogen. Then the powder was re-suspended in a lysis buffer (8 mol L-1urea, 4%CHAPS, 40 mmol L-1Tris-HCl), with a final concentration of 1 mmol L-1phenylmethylsulfonyl fluoride (PMSF) and 2 mmol L-1ethylenediaminetetraacetic acid (EDTA). After 5 min of vortex, 10 mmol L-1dithiothreitol (DTT; final concentration) was added to the solution. After sonication for 15 min, the samples were centrifuged for 20 min at 25 000×g,and the supernatant was mixed well with 10 mmol L-1DTT for 1 h at 56°C. Then iodoacetamide (IAM) was added to a final concentration of 55 mmol L-1and reacted in the dark for 45 min. The solution was mixed well with ice-cold acetone(1:4, v/v) at -20°C for 2 h. After 20 min of centrifugation at 25 000×g, the supernatant was discarded and the residual material was sonicated for 15 min until dissolved in 200 μL of 0.5 mol L-1triethylammonium bicarbonate (TEAB) solution.Finally, after being centrifuged at 25 000×g for 20 min, the supernatant was collected and protein was quantified by the Bradford method (Kruger 1998). For digestion, 100 μg of protein of each sample was weighed and then digested with trypsin at 37°C for 12 h (Sigma; 1:20 w/w added at 0 and 4 h).
2.4. iTRAQ labeling and LC-MS/MS proteomic analysis
The iTRAQ labeling of peptide samples derived from the control and the Cd-treated conditions were performed using an iTRAQ reagent 8-plex kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. We performed two biological replications at four different time points, with a total of 16 samples prepared for quantitative proteomics experiments. The control samples taken at 12,24, 48, and 72 h were each labeled with iTRAQ reagents with molecular masses of 113 (12 h), 114 (24 h), 115 (48 h), and 116 (72 h) Da, respectively, while the Cd-treated samples were labeled with 117 (12 h), 118 (24 h), 119 (48 h), and 121 (72 h) Da iTRAQ tags, respectively. After labeling,all samples were pooled and purified with a strong cation exchange chromatography (SCX) column using an Agilent 1200 High Performance Liquid Chromatography(HPLC) System (Agilent, USA) and separated by liquid chromatography (LC) using the Eksigent nanoLC-Ultra 2D System (AB SCIEX, USA). Mass spectrometer data acquisition was performed with a Triple TOF 5600 System(AB SCIEX, Concord, ON) fitted with a Nanospray III source(AB SCIEX, Concord, ON) and a pulled quartz tip as the emitter (New Objectives, Woburn, MA).
2.5. Proteins identification and quantification
MS/MS spectra were searched using MASCOT engine Software (Matrix Science, London, UK; version 2.2)embedded in Proteome Discoverer 1.4 Software (Thermo Electron, San Jose, CA), and run against the maize protein database (http://ftp.maizegdb.org/MaizeGDB/FTP/maize_proteome/proteome.fasta). For protein identification,the following options were used: peptide mass tolerance=±0.05 Da, fragment mass tolerance=0.1 Da, enzyme=trypsin,missed values=monoisotopic, max missed cleavage=1,Fixed modification: carbamidomethyl (C), iTRAQ&8plex(N-term), iTRAQ&8plex (K), and variable modification:iTRAQ&8plex (Y) (Sandberg et al. 2012).
DAPs, including significantly down- or up-regulated proteins, were identified using the ProteinPilot Software(AB SCIEX, USA). The expression ratios were used to assess fold changes in the abundance of the proteins identified in the Cd-treated plants vs. the control plants.Duncan’s multiple range test was used to identify significant differences (P<0.05) in means between the Cd-treated and control plants. Confidence scores >1.5-fold (increased abundance) or <0.67-fold (decreased abundance) were used as the qualification criteria (Sang et al. 2016), which corresponded to a peptide confidence level of 95% (Yang et al. 2013). All data were normalized by bias correction,which is an algorithm in ProteinPilot that corrects for unequal mixtures, combining the different labeled samples. DAPs were defined as those showing at least a 1.5-fold change relative to their corresponding controls (Rogers et al. 2012).
2.6. Quantitative real-time PCR for selected gene expression profiles
To check the correlation between RNA expression and protein expression, nine DAPs which were detected by three time points, were used to perform qPCR analysis. TRIZOL Reagent (Invitrogen, Carlsbad, CA) was used to extract total RNA from frozen root tissue according to the manufacturer’s protocol. Total RNA (1 μg) was reverse-transcribed using PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa Corp., Dalian, China). qPCR was performed using SYBR Premix Ex Taq (TaKaRa Corp., Dalian, China) and the StepOne Plus Real-time PCR System (Applied Biosystems,Foster City, CA, USA). All PCR primers (Appendix A)were designed using Primer 5.0 and primer specificity was determined by both agarose gel electrophoresis and melting curve analysis. The relative expression levels of target genes were estimated using the formula 2-ΔΔCtmethod (Livak and Schmittgen 2001). gpn1 was used as internal reference for quantifying mRNA levels. Three independent biological replicates were analyzed for each sample.
2.7. Protein interaction network analysis
Gene Ontology (GO) functional categories were assigned to DAPs, using the web-based agriGO tools (http://bioinfo.cau.edu.cn/agriGO/). KOBAS Software (Xie et al. 2011) was used to perform pathway enrichment analysis by testing the statistical enrichment of DAPs in the Kyoto encyclopedia of genes and genomes (KEGG) pathways. Protein-protein interaction networks were analyzed by the publicly available program STRING (http://string-db.org/).
3. Results
3.1. Cd treatment effects on maize root and Cd accumulation characteristics
In order to have an initial perspective of the strategies triggered to decrease Cd toxicity, we assessed the phenotype of plants exposed to Cd for different time points.Maize plants grown under Cd stress showed a significant reduction of the length of both roots and shoots (Fig. 1-A-E).In the controls, the length of both roots and shoots increased linearly over a 72-h period. However, when subjected to Cd, it seemed that the maize growth was restrained, no significant difference was observed in either roots or shoots over the first 24 h, and a little difference was observed between 48 and 72 h. At the end of the experimental period(72 h), the leaves of Cd-treated plants showed symptoms of chlorosis, and necrotic areas began to be evident at their tips. After 48 h of Cd treatment, Cd content in roots increased significantly between Cd treatment and control groups, with a difference of 45.23- and 84.24-fold in Jing724 and B73, respectively (Fig. 1-F and G). Therefore, this suggested that Jing724 was relatively more resistant to Cd stress than B73.
3.2. Overview of the identified maize root proteins
Comparisons between the two biological replicates showed good analytical reproducibility (Appendix B), with a total of 2 573 and 2 440 proteins (Jing724) detected at a false discovery rate (FDR) of 5% (Appendix B). We considered those proteins with both a fold change of >1.5 and a P-value<0.05 as DAPs (Yang et al. 2013; Hu et al. 2015a).Considering two replicates together, there were 386, 181,236, and 242 up-regulated DAPs at 12, 24, 48, and 72 h of Cd treatment, respectively. Meanwhile, we detected 347,126, 263, and 334 down-regulated DAPs at each of the four time points, respectively (Table 1). Comparison of upregulated DAPs among different developing stages revealed 59, 9, 48, 6, 11, and 30 DAPs constantly up-regulated at 12&24 h, 12&48 h, 12&72 h, 24&48 h, 24&72 h, and 48&72 h, respectively. By contrast, there were 22, 3, 65,11, 7, and 58 DAPs constantly down-regulated at 12&24 h,12&48 h, 12&72 h, 24&48 h, 24&72 h, and 48&72 h,respectively (Appendix C). The levels of a total of 14 proteins were up- (3) or down-regulated (11) at three of the four time points (Table 2). We considered these proteins identified at two or three different time points as candidate regulated proteins or super candidate regulated proteins, respectively.
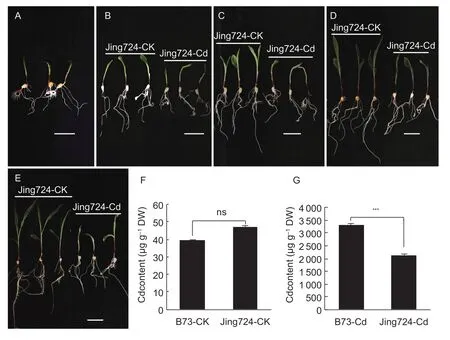
Fig. 1 Effects of cadmium (Cd) stress on maize seedling growth. A-E, samples at 0, 12, 24, 48, and 72 h, respectively, scale bar=5 cm. F and G, Cd contents accumulated at control and Cd-treatment conditions at 48 h, respectively. ns, no sigificant difference;***, significant difference level at P<0.0001. Bars mean SD.
To investigate the functions of these DAPs, we further classified the 166 up-regulated and 177 down-regulated candidate regulated proteins and super candidate regulated proteins into 123 up-regulated and 95 down-regulated categories based on their GO annotations (Appendices D and E). GO enrichment of the candidate regulated proteins and super candidate regulated proteins revealed that 76, 29, and 121 protein categories were enriched in the biological process (BP), molecular function (MF), and cellular component (CC) categories, respectively. For these up-regulated categories, translation, structural constituent of ribosome, and cytosolic ribosome were the most abundant categories in BP, MF, and CC, respectively. Among the down-regulated protein categories, response to stimulus,oxidoreductase activity, and plasma membrane were the most abundant categories in BP, MF, and CC, respectively.
3.3. Gene annotations of super candidate regulated proteins
A total of 14 proteins had higher (3) or lower levels (11)at three different time points (Table 2), indicating that those proteins were likely to be responsive to Cd stress.GRMZM2G081652 is a transport protein, GRMZM2G158034 is the ribosomal protein eukaryotic 40S subunit S13 protein,the protein abundance was significantly higher than 0 h after 12, 24, and 72 h of Cd treatments. GRMZM2G308687, a glutathione S-transferase, which is best known for its ability to catalyze the conjugation of the reduced glutathione to xenobiotic substrates for detoxification, was more abundant after 12, 48, and 72 h of Cd treatment. Among the seven less abundant proteins after 12, 24, and 72 h of Cd treatment, we identified a stress biotic and pathogenesisrelated thaumatin-like protein (GRMZM2G039639), a cell organization protein (GRMZM2G047055), a lipid metabolism protein (GRMZM2G059693), two sugar transporters(GRMZM2G146670 and GRMZM2G150616), a jasmonate synthesis degradation lipoxygenase (GRMZM2G156861),and a protein with unknown function (GRMZM2G161746).Four proteins were down-regulated at 24, 48, and 72 h,that is, GRMZM2G049930, GRMZM2G071390,GRMZM2G178817, germin-like stress abioic related proteins, and GRMZM2G156684, a cell organization protein.
3.4. Validation of DAPs by qPCR
We selected nine super candidate regulated proteins for transcriptional analysis in order to confirm whether gene expression data correlated with protein expression (Fig. 2).qPCR analysis indicated that the transcriptions patterns of GRMZM2G047055, GRMZM2G156684, GRMZM2G059693,GRMZM2G146670, and GRMZM2G156861 follow the same trend as our proteomic data. However, the transcription data GRMZM2G158034, GRMZM2G308687,GRMZM2G150616, and GRMZM2G039639 did not match our data on protein abundance. The different trends between RNA expression and protein levels could also be acceptable,since they do not have inevitability correlation (Wang et al.2010). Post-translation modification, translational or posttranslational regulation may cause a discrepancy between the transcriptional patterns and proteomics observations(Washburn et al. 2003).
3.5. Pathways differentially regulated under Cd treatment
KEGG pathway enrichment revealed that pathways with significant changes in response to Cd stress fell into 35 up-regulated and 27 down-regulated categories. Ribosome and proteasome pathways were significantly up-regulated,with a corrected P-value lower than 0.05. The ribosome pathway displayed the largest rich factor and protein number, with a P-value of 3.71×10-24(Fig. 3). Though glutathione metabolism pathway did not reach our threshold for significance, it showed up-regulation under Cd stress.The top three down-regulated pathways, phenylpropanoid biosynthesis, phenylalanine metabolism, and endocytosis,were significantly differentially expressed with a corrected P-value<0.05 (Appendix E). The phenylalanine metabolism pathway is commonly involved in Cd stress responses in crops (Wang et al. 2017). These results indicated that the five pathways related to ribosome, proteasome,phenylpropanoid biosynthesis, phenylalanine metabolism,and endocytosis, play a vital role in maize root during exposure to Cd stress.

Table 1 Summary of differentially regulated proteins at various time points of cadmium (Cd) treatments1)
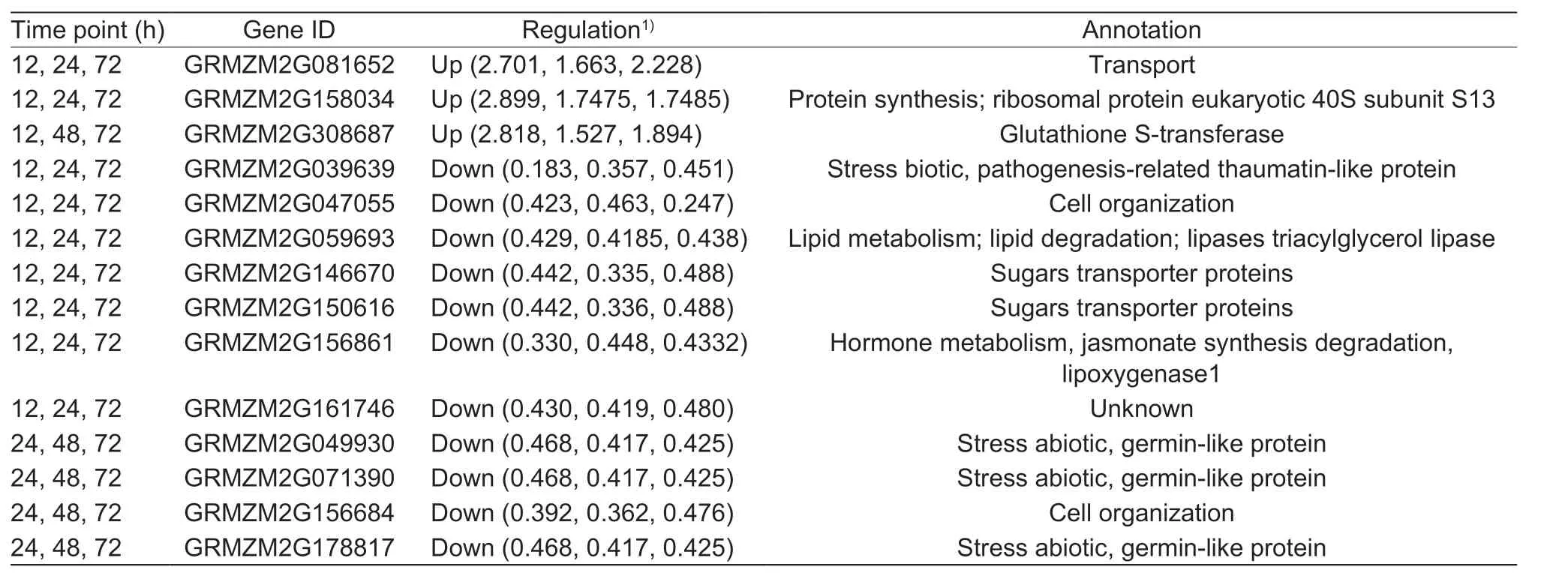
Table 2 Summary of proteins that were regulated at three time points in response to cadmium (Cd) treatment

Fig. 2 Expression analysis of differentially abundant proteins at 24 or 48 h using RT-qPCR. The expression level of Jing724-CK in maize root at 24 h (GRMZM2G158034, GRMZM2G039639, GRMZM2G047055, GRMZM2G059693, GRMZM2G146670,GRMZM2G150616, GRMZM2G156861) and 48 h (GRMZM2G308687 and GRMZM2G156684) was set as “1.0”. Different letters above the bars indicate a significant difference at P<0.05. Bars mean SD.

Fig. 3 Summary of all candidate regulated proteins. A, heat map of candidate regulated proteins by log2(fold change). G1-3,three main clusters which have different protein abundant pattern based on hcluster. B, KEGG pathway enrichment. The Y-axis is different pathways, the X-axis is the number of rich factors, circle size means the protein number, P-value is the significant level,the larger the -log10P-value is, the more significant the pathway is related to Cd stress.
4. Discussion
Cadmium toxicity often leads to decreased root length and dry mass (Gratão et al. 2009). Here, we observed a severe reduction in both root and shoot growth after a 72 h-period Cd treatment on maize seedlings. The Cd concentrations in the roots of B73 (3 306.47 μg g-1) seedlings was significantly higher than that of Jing724 (2 127.61 μg g-1) after a 48-h Cd treatment, suggesting that Jing724 plants can adapt to Cd stress conditions better than B73(Fig. 1-G). We used Jing724 for the following quantitative proteomic study, identified and quantified numerous DAPs via an iTRAQ-based quantitative proteomic approach. In general, the abundance of DAPs with functions related to protein degradation, cellular component assembly,nucleosome and chromatin assembly, and ribosome protein synthesis increased substantially, while the levels of proteins involved in ion binding, antioxidant, energy, and transport were reduced significantly. These proteins were assigned to different functional categories and the following discussion is based on such functional classification and related physiological responses.
4.1. Proteins involved in protein modification and post-translational modifications
To overcome the stress of environmental stimuli, it is of great importance to coordinate the balance between protein synthesis and degradation (Hinkson and Elias 2017). In the present study, we identified 70 differentially accumulated proteins implicated in protein synthesis, modification, and degradation (Fig. 4). Twenty-eight of 33 protein synthesis ribosomal proteins accumulated higher levels under Cd stress, indicating that Cd stress may induce an overall up regulation of protein synthesis to balance plant growth and Cd tolerance. Similar up-regulation of ribosome proteins were found in the hyperaccumulator maize line 178 (Shen et al. 2013), radish root (Wang Y et al. 2016), and Chinese flowering cabbage (Wang et al. 2017) under Pb stress conditions. However, the down regulation of ribosomal proteins is also observed in many studies. The abundance of ribosomal proteins in both sensitive and tolerant rice cultivars decreases significantly after aluminum treatment(Wang et al. 2014). Additionally, the abundance of ribosomal proteins decreases in salt-treated Jing724 (Luo et al. 2017),implying salinity may induce an overall down-regulation of protein synthesis in this maize line. The abundance levels for six posttranslational modification proteins(GRMZM2G043383, GRMZM2G070542, AC206425.3,AC209208.3, GRMZM2G046297, and GRMZM2G125762)were observed to be down-regulated, and 12 of 15 protein degradation family species (GRMZM2G012160,GRMZM2G438551, GRMZM2G038636, GRMZM2G000601,GRMZM2G173756, GRMZM2G053764, GRMZM2G146374,GRMZM2G056870, GRMZM2G147671, GRMZM2G165926,GRMZM2G467059, and GRMZM2G053898) were shown to be up-regulated under Cd exposures. These data above implied a sophisticated network of protein modification under Cd stress conditions, Cd stress accelerated both protein synthesis and degradation, mechanisms in protein balance need to be further investigated. The abundance of 13 histone proteins were increased under Cd stress, which can undergo several different types of post-translational modifications that affect transcription, DNA repair, DNA replication, and chromosomal stability. This result is in accordance with previous studies that reported the modifications of chromatin structure of the N-terminal tails of histones, implying that hypomethylation or hypermethylation of histone proteins,play a vital role in post-regulation of Cd-stress responsive genes in maize seedling roots (Chinnusamy et al. 2008;Chinnusamy and Zhu 2009).
4.2. Proteins involved in ROS homeostasis and defense
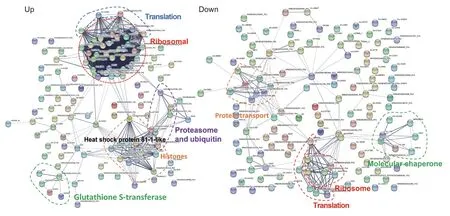
Fig. 4 The protein-protein interaction network among differentially abundant proteins (DAPs) in maize seedling roots under cadmium(Cd) stress. Left, up-regulated; right, down-regulated.
Cadmium exposure sharply increases H2O2production,and plants continuously generate ROS under stress conditions (Yang et al. 2016), which not only damage cellular components, but also are important for signaling regulating many biological processes (Foyer and Noctor 2005). An increase in the abundance of defense proteins related to ROS scavenging and molecular chaperones for re-establishing normal protein conformation facilitates to the maintenance of redox homeostasis in heavy metal stressed plants (Hossain and Komatsu 2012). In this study, the abundance level changes for many antioxidant enzymes were increased,including 13 peroxidases (AC205413.4, GRMZM2G108077,GRMZM2G023840, GRMZM2G085967, GRMZM2G138450,GRMZM2G027217, GRMZM2G437207, GRMZM2G427937,GRMZM2G427954, GRMZM2G504757, GRMZM2G065585,GRMZM2G097207, and GRMZM2G120962), and five glutathione S-transferase (GRMZM2G308687,GRMZM2G162486, GRMZM2G035502, GRMZM2G144153,and GRMZM5G855672). Among them, an antioxidant enzymes AC205413.4 is peroxidase (POD), which can detoxify H2O2by oxidizing ascorbate, protect the cell membrane from hydroxyl radical-induced lipid peroxidation(Barber and Thomas 1978). GRMZM2G308687, a GSTlike protein that is involved in multifunctional activities in phytochelatin (PC) synthesis, methylglyoxal detoxification,and ROS scavenging through the ascorbate-GSH cycle(Nocito et al. 2002; Hossain and Komatsu 2012). This was in agreement with the result concluded by Tuomainen et al.(2006), proteins controlling ROS scavenging are particularly abundant in root tissues of two accessions of the Cd-zinc hyperaccumulator Thlaspi caerulescens. At the early stages of maize grain development, Yu et al. (2016) identified 58 DEPs as ROS related proteins, such as dehydroascorbate reductase, glutathione S-transferase (GST), superoxide dismutase, and thioredoxin. In addition, we identified six candidate regulated proteins conditioning abiotic and biotic stress response, including GRMZM2G112165 (a heat shock protein and a type of molecular chaperon),GRMZM2G062373, GRMZM2G106690 (universal stress protein (USP) family members), AC205274.3,GRMZM2G304442, and GRMZM2G159503 (pathogenesisrelated (PR) proteins). It is worthy to note that the molecular chaperon GRMZM2G112165 provides a joint among ribosome, histone, and glutathione S-transferase proteins. Moreover, we found proteins related to glutathione metabolism to be up-regulated, suggesting that this pathway plays an important role in Cd detoxification (Figs. 3-B and 4).
4.3. Proteins involved in signal transduction and cell wall degradation
Cell wall is the first barrier to invade a plant cell, many stress-responsive signaling proteins anchored to the cell wall or the membrane were activated when the outer stress conditions are perceived (Jamet et al. 2006). Activation of phosphorylation cascades, Ca-calmodulin system,ROS signaling, and stress-related hormones eventually converge regulating transcription factors that are deputed to the activation of gene sets responsible for response to stress (DalCorso et al. 2010). In the current study, the abundance of two calcium signaling (GRMZM2G113453 and AC202073.4) proteins, and one mitogen-activated protein kinase GRMZM2G163709 was increased under Cd stress, indicating that calcium and MAPK are thought to be involved in Cd signaling to the nucleus, followed the activation of Cd response proteins. Moreover, the levels of four cell wall degradation related proteins,GRMZM2G001514, GRMZM2G021794 (arabinogalactan proteins, AGPs; fasciclin-like protein), GRMZM2G035503 and GRMZM2G136895 (mannan-xylose-arabinose-fucose),and GRMZM2G107073 and GRMZM2G088531 (pectate lyases and polygalacturonases) were reduced after Cd treatment. A previous study reported lower levels of proteins involved in cell wall and cytoskeleton constitution were also observed in Mn-treated Citrus sinensis roots (You et al.2014), implying that protecting the cell wall integrity from heavy metal is vital for signal transduction and heavy metal tolerance.
4.4. Proteins involved in carbohydrate and energy metabolism
Several protein species involved in carbohydrate and energy metabolism-related pathways, including TCA cycle (AC234171.1 and GRMZM2G064695), respiratory electron transport (GRMZM2G015401, GRMZM2G152827,GRMZM2G050003, and GRMZM2G064473) and carbon metabolism (GRMZM2G065585, GRMZM2G097207,GRMZM2G120962, AC217401.3, GRMZM2G008247, and GRMZM2G172369) were shown to be repressed under Cd exposure. This was in accordance with the result of You et al. (2014), that proposed that the down-regulation of proteins abundance in Mn treated C. sinensis roots with lower accumulation of carbohydrates may provide an advantage to the net carbon balance by lowering related metabolic processes, enhanced the Mn tolerance in C. sinensis. Moreover, waterlogging stress also represses proteins functioning in sucrose metabolism in maize seedling roots (Yu et al. 2014). On the contrary, to maintain ion homeostasis, or at least to protect cells against excess metals, plants activate mechanisms producing energy, such as ATPase and proton pumps (Martinoia et al. 2007). The abundance of some of the proteins involved in carbohydrate metabolism increases after upon Pb(NO3)2at 500 mg L611(Pb500) exposure, but decrease under 1 000 mg L611(Pb1000) (Wang Y et al. 2016). In addition, the transport of Cd complexes into the vacuole or in the apoplast is also dependent on ATP membrane pumps (Dalcorso et al. 2010).V-ATPase activity might play a central role in response to excess zinc (Fukao et al. 2011). ATPase3 is the primary factor in stress responses to Cd variation in Arabidopsis thaliana leaves (Chao et al. 2012). Previous studies also proposed that the up-regulation of proteins related to energy metabolism imposes an enhanced energy demand(Visioli and Marmiroli 2013). However, in the current study,the abundance of two proteins (GRMZM2G014240 and GRMZM2G170927) with the H_PPase domain was repressed under Cd stress. Moreover, the abundance level changes of four proteins (GRMZM2G015401, GRMZM2G152827,GRMZM2G050003, and GRMZM2G064473) regarding to metabolite transporters at the mitochondrial membrane were decreased under Cd exposure. These results above implied that the energy metabolism pathway was seriously damaged under Cd stress, alternative pathways, such as protein modification, histone modification may play a role in Cd tolerance.
5. Conclusion
Through the exposure of maize seedling roots to Cd, the growth of both the roots and shoots were seriously inhibited during the first 72 h. There are 733, 307, 499, and 576 DAPs isolated at 12, 24, 48, and 72 h, respectively. It is a remarkable fact that these high-level DAPs are enriched in translation, structural constitute of ribosome, cytosolic ribosome categories. On the contrary, those low-level DAPs are enriched in response to stimulus, oxidoreductase activity,and plasma membrane. Fourteen proteins have continuously higher or lower levels at three of the four time points. Among them, the glutathione S-transferase GRMZM2G308687 is more abundant after 12, 48, and 72 h Cd treatment, which is a key protein involved in phytochelatin synthesis for Cd stress tolerance. Ribosome pathway, enriched by high level DAPs, is also involved Pb stress, the mechanism needs to be further investigated. All in all, together with proteins involved in protein and post transcriptional modification,ROS homeostasis, carbohydrate and energy metabolism,the proteome changes gave insights into the mechanisms of both Cd stress damage and Cd tolerance in maize seedling roots.
Acknowledgements
The authors would like to thank Li Ting, Chen Hao,and Yang Fengling (Beijing Academy of Agriculture &Forestry Sciences), for their help with data processing and experiments performing. This work was supported by the Foundation for Young Scientist of Beijing Academy of Agriculture & Forestry Sciences, China (QNJJ201505) and the National Key Research and Development Program of China (SQ2016ZY03002163).
Appendicesassociated with this paper can be available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
 Journal of Integrative Agriculture2019年10期
Journal of Integrative Agriculture2019年10期
- Journal of Integrative Agriculture的其它文章
- Application of virus-induced gene silencing for identification of FHB resistant genes
- Strategies to enhance cottonseed oil contents and reshape fatty acid profile employing different breeding and genetic engineering approaches
- Maize/peanut intercropping increases photosynthetic characteristics,13C-photosynthate distribution, and grain yield of summer maize
- Rhizosphere soil bacterial community composition in soybean genotypes and feedback to soil P availability
- Effect of biochar on grain yield and leaf photosynthetic physiology of soybean cultivars with different phosphorus efficiencies
- lnheritance of steroidal glycoalkaloids in potato tuber flesh