
脱泛素酶OTUD7B调控肿瘤微环境和肿瘤进程的分子机制*
2019-09-23 01:07林丹丹史建红
中国病理生理杂志 2019年9期
林丹丹, 申 阳, 解 欣, 2, 史建红△
(1河北大学附属医院实验中心, 河北省肿瘤放化疗机制与规程研究重点实验室, 2河北大学医学院, 河北 保定 071000)
泛素化(ubiquitination)是一种蛋白质翻译后修饰过程,泛素(ubiquitin)以共价连接的方式形成多聚泛素链并结合到靶蛋白赖氨酸(lysine,K)残基上,该过程由泛素激活酶(ubiquitin-activating enzymes,UBE,E1)、泛素结合酶(ubiquitin-conjugating enzymes,UCE,E2)和泛素连接酶(ubiquitin ligases,E3)顺序激活[1]。经典的泛素化调节方式是通过泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)介导蛋白质降解而实现对靶蛋白含量的精确调控[2-3]。随着人们对泛素化认识的不断深入,除了UPS介导的蛋白质降解和更新外,泛素化还可通过非降解机制调节细胞功能,如K63-和M1 (Met1)-多聚泛素链可形成细胞内反应的平台,参与激酶活化和细胞内信号转导[4-5]。泛素化是一个可逆的动态过程,受到E3和脱泛素酶(deubiquitinase,DUB)的共同调节[6]。OTUD7B(OTU domain-containing 7B)是一种多功能脱泛素酶,介导K11-、K48-和K63-多聚泛素链解离,在生长发育、免疫应答和恶性肿瘤等生理病理过程中发挥重要作用。
1 OTUD7B与脱泛素调节
1.1OTUD7B的基因结构和特点 OTUD7B是OTU脱泛素酶超家族成员。通过晶体结构分析发现,OUT超家族蛋白在氨基酸序列上与其他类的脱泛素化酶有较大差异,该家族蛋白核心区域由2个螺旋结构和5个β-折叠组成。不同的OTU 家族成员其OTU结构域大小不同,但均已被证实具有脱泛素酶活性[7-8]。OTU家族的共同结构特点为:含有OTU结构域、泛素相关(ubiquitin-associated,UBA)结构域、Npl4锌指(Npl4 zinc finger)结构域和 A20样锌指(A20-like zinc finger)结构域。
OTUD7B的分子结构包括 UBA、OTU、cytadhesin P30/P32和A20样锌指结构域,见图1。现已知UBA结构域不直接影响OTUD7B的脱泛素酶活性,但可介导K63-多聚泛素链解离,影响NF-κB信号通路[9];OTU结构域发挥主要的DUB活性[10];cytadhesin P30/P32结构域参与细胞黏附;A20样锌指结构域参与泛素链结合。
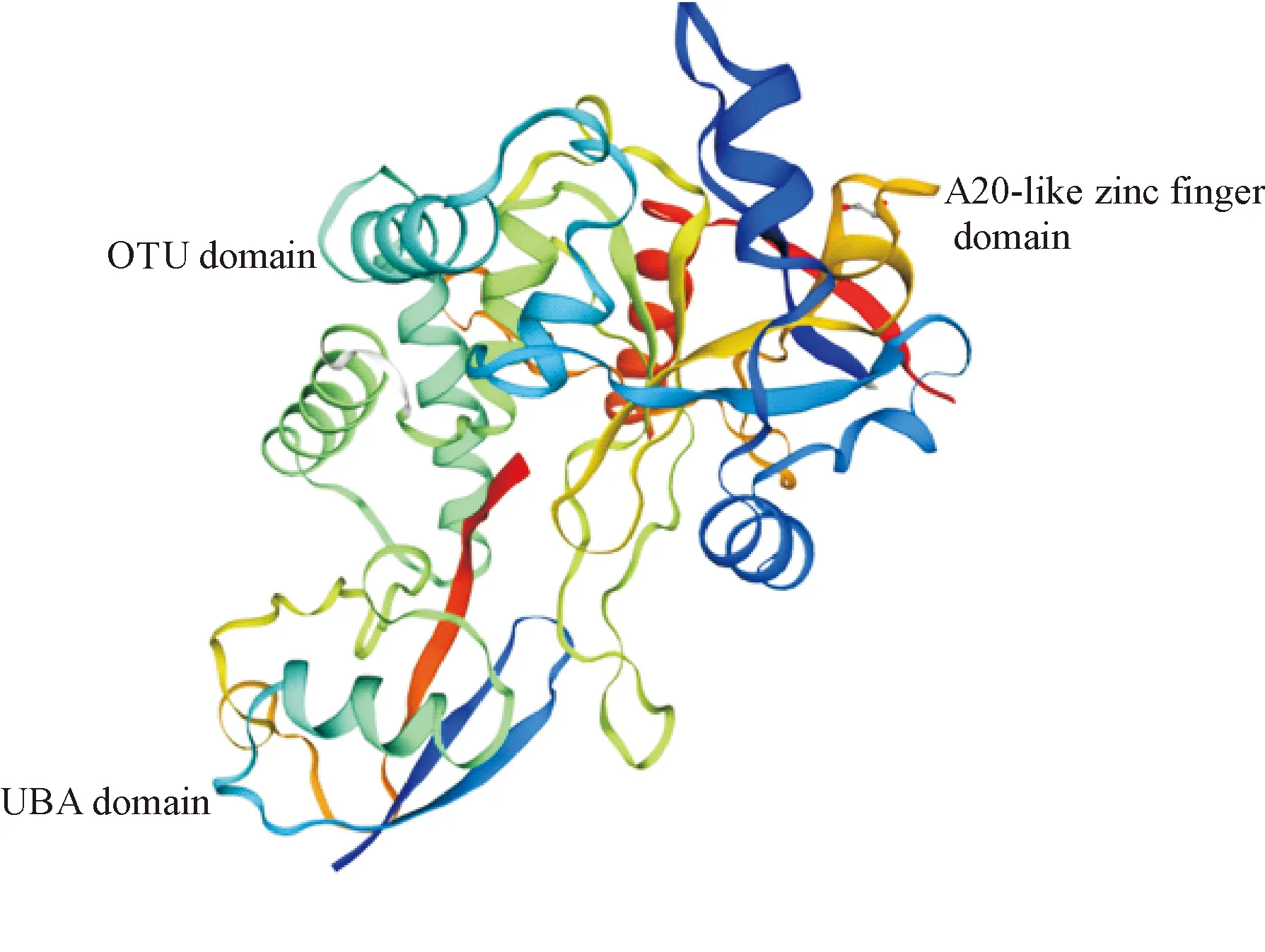
Figure 1.The structure of OTUD7B.
图1 OTUD7B分子结构示意图
1.2OTUD7B催化K11-、K48-和K63-多聚泛素链水解 根据泛素连接方式不同,多聚泛素链共有8种类型:K6-、K11-、K27-、K29-、K33-、K48-、K63-和M1-多聚泛素链。K48-泛素化主要介导靶蛋白通过UPS降解,调节细胞内蛋白质的稳定性;K63-泛素链通过非降解机制介导细胞内信号传递;K11-多聚泛素链的功能研究目前尚不十分清楚。体外实验证实,OTUD7B可特异性靶向K11-多聚泛素链,催化K11-脱泛素化过程[11]。晶体结构分析显示,K11-泛素链可结合OTUD7B的酶催化S1位点OTUD7B[12]。OTUD7B通过调节HIF-1α分子K11-脱泛素化,抑制缺氧诱导因子1α(hypoxia-inducible factor-1α,HIF-1α)通过UPS非依赖途径降解[13]。OTUD7B也可与UBE2S/E2-EPF和UBE2C/UbcH10共同调节有丝分裂后期促进复合体(anaphase-promoting complex,APC/C)的K11-多聚泛素链的脱泛素化和泛素化过程,影响其稳定性,参与细胞周期调控和内质网相关降解(endoplasmic reticulum-associated degradation,ERAD)[14-17],见图2。
尽管体外实验和晶体实验数据显示,与其他7种泛素链相比,OTUD7B更容易结合并水解K11-多聚泛素链,一些细胞实验证实OTUD7B也可靶向细胞内蛋白的K48-和K63-多聚泛素链,促进靶蛋白脱泛素化并调节细胞信号转导[18]。OTUD7B还可直接作用于E3连接酶,催化肿瘤坏死因子受体相关因子3(tumor necrosis factor receptor-associated factor 3,TRAF3)的K48-多聚泛素链解离,抑制TRAF3通过UPS降解[10]。在低氧条件下,OTUD7B可以诱导TRAF6分子K63-脱泛素化,调节NF-κB活化。TNFα可刺激OTUD7B募集到TNFR并催化K63-多聚泛素链与RIP解离[18]。OTUD7B也可以使GβL发生K63-脱泛素化,促进哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)复合体2(mTOR complex 2, mTORC2)的稳态和活性[19],见图2。
2 OTUD7B与肿瘤的发生发展
2.1OTUD7B诱导乳腺癌增殖和耐药 OTUD7B在不同肿瘤中的作用不尽相同。OTUD7B在乳腺癌中高表达,并被证实与乳腺癌高侵袭性相关[20]。OTUD7B可催化乳腺癌细胞的表皮生长因子受体(epidermal growth factor receptor,EGFR)脱泛素化,抑制EGFR通过UPS降解,促进乳腺癌细胞增殖和存活[20]。在三阴性乳腺癌(triple-negative breast can-cer,TNBC)中,OTUD7B可通过负调节NF-κB-Lin轴,导致微小RNA(microRNA,miRNA,miR)let-7介导的caspase-3下调,促进乳腺癌高侵袭性和紫杉醇耐药[21]。
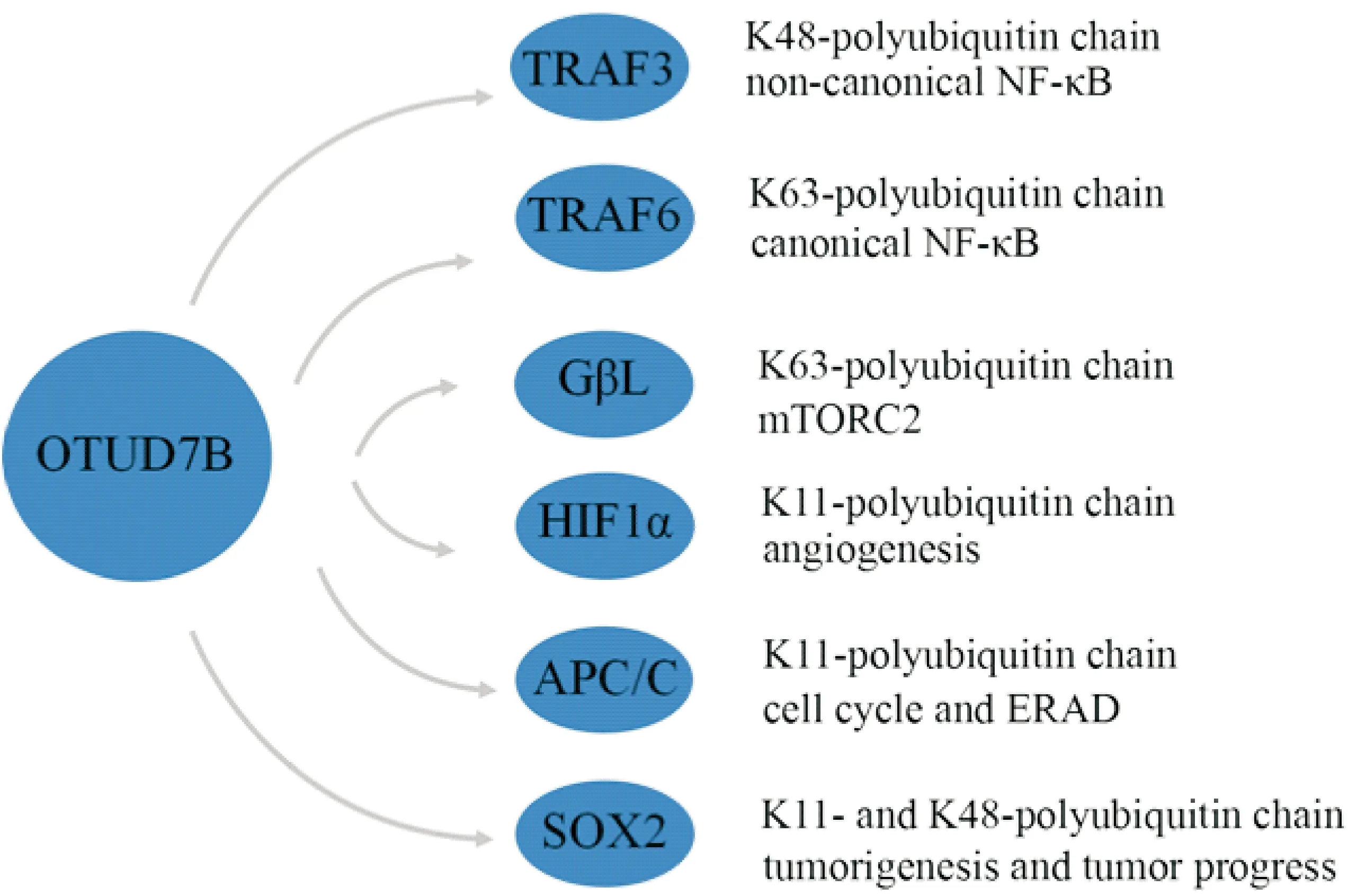
Figure 2.OTUD7B catalyzes deubiquitination.
图2 OTUD7B催化的脱泛素化过程
2.2OTUD7B在肝癌中的作用 与乳腺癌不同,在肝细胞癌(hepatocellular carcinoma,HCC)的研究发现,OTUD7B在HCC的表达水平明显低于癌旁肝组织。OTUD7B水平越低的肝癌患者,其肿瘤越大、临床分期越高、组织分化程度往往较低并伴有淋巴结转移,血管侵袭能力增加,伴有早期复发,预后较差[22]。OTUD7B可通过诱导上皮-间充质转化,抑制肝癌细胞的迁移和侵袭[23]。
2.3OTUD7B与肺癌 OTUD7B在肺癌中的表达和作用机制目前仍不十分清楚。有研究显示,OTUD7B抑制H157非小细胞肺癌细胞株的NF-κB核转位发挥抑癌作用,参与紫杉醇和MEK抑制剂U0126联合作用诱导的H157细胞死亡[24]。OTUD7B基因敲除小鼠用KRAS诱导的自发性肺癌发生率显著下降[19]。OTUD7B在肺腺癌组织中高表达,OTUD7B表达水平与肺癌的不良预后呈正相关[25]。
2.4OTUD7B与其他肿瘤 OTUD7B在其他肿瘤中的作用尚未明确,但有研究表明,OTUD7B通过其脱泛素化修饰,调节性别决定区Y框蛋白2(sex determining region Y-box 2,SOX2)的稳定性[26]。SOX2在乳腺癌、肺癌、胃肠道肿瘤、皮肤肿瘤、前列腺癌、中枢神经系统等多种类型肿瘤中表达均上调,参与肿瘤生长、转移等过程,与不良预后及化疗耐药相关,因此,OTUD7B可能通过调节SOX2的稳定性调控多种类型肿瘤进程[27]。
3 OTUD7B对肿瘤功能的调控
3.1促进肿瘤生长和转移 EGFR信号活化过程往往伴随一些E3泛素酶,如CBL,迅速结合活化的EGFR并介导EGFR通过UPS降解,从而快速终止EGFR信号的持续活化[28]。OTUD7B通过其OTU结构域催化EGFR脱泛素化,抑制EGFR降解,促进EGFR下游分子持续活化[20]。在宫颈癌HeLa细胞和乳腺癌MDA-MB-468细胞中,EGF刺激OTUD7B与EFGR结合并抑制EGFR泛素化降解,增强乳腺癌细胞增殖和迁移,促进肿瘤的生长和侵袭[20]。
胰岛素样生长因子受体(insuline-like growth factor receptor,IGFR)激活下游磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)-丝氨酸/苏氨酸蛋白激酶(protein kinase B,PKB,即AKT) 1-mTOR通路和MAPK通路,调节肿瘤细胞增殖、凋亡和存活。IGFR泛素化和脱泛素化的动态平衡对IGF信号途径的触发和终止调节也至关重要[29]。肺腺癌组织中,OTUD7B和IGFR表达呈正相关[25],提示OTUD7B可能是调节IGFR信号通路的一种重要分子,其分子作用机制尚待进一步研究。
转录因子SOX2主要经Shh、Wnt、成纤维细胞生长因子受体(fibroblast growth factor receptors,FGFR)和转化生长因子β(transforming growth factor beta,TGF-β)信号通路活化,诱导下游促细胞存活、生长、增殖基因的转录[30]。泛素连接酶和脱泛素酶共同调节SOX2的稳定性,从而影响肿瘤干细胞的特性,促进多种肿瘤的生长、侵袭和转移能力,影响预后转归[26]。
3.2促进肿瘤发生 mTOR是细胞生长和多条代谢途径调节的关键分子。mTOR可形成2种复合物:mTORC1和mTORC2[31-32]。mTORC1对雷帕霉素敏感,能磷酸化S6K1和4E-BP1促进蛋白质翻译[33];mTORC2主要响应细胞外生长因子信号通路,如PI3K信号。OTUD7B通过靶向mTORC1和mTORC2共有成分GβL分子K63-多聚泛素链,催化GβL脱泛素化,引发mTORC1解离,促进mTORC2形成,激活下游Akt信号,促进肿瘤发生[19]。
3.3细胞周期的调节 细胞分裂增殖的过程中,APC/C作为一种E3连接酶,促进有丝分裂细胞周期蛋白的降解,调节周期依赖性蛋白激酶(cyclin-dependent kinase,CDK)活性,控制细胞周期依赖性钙振荡(cell cycle-dependent calcium oscillation)[15, 34]。OTUD7B可拮抗APC/C的泛素化作用,催化K11-多聚泛素链解离,抑制APC/C介导的有丝分裂进程和微核形成,抑制肿瘤细胞增殖[16]。
4 OTUD7B调节肿瘤微环境
4.1经典NF-KB信号影响肿瘤发生发展 泛素化和脱泛素化调控在NF-κB信号激活的多个环节中发挥调节作用[4, 35-37]。非小细胞肺癌细胞株H157中,OTUD7B抑制NF-κB核转位,降低IL-8和ICAM-1表达。耐药基因DJ-1结合OTUD7B并抑制其脱泛素酶活性[24]。在缺氧条件下,OTUD7B可抑制RelA磷酸化和TRAF6泛素化,抑制NF-κB核转位,减少缺血再灌注反应中的炎症和损伤[38]。TNF-α诱导HEK293细胞和人脐静脉血管内皮细胞中OTUD7B表达,OTUD7B通过其OTU结构域催化K48-和K63-脱泛素化,负反馈抑制TNFα诱导的IKK活化、IκBα磷酸化降解和NF-κB核转位[18]。
4.2非经典NF-κB信号调节免疫反应 与经典NF-κB不同,非经典NF-κB信号途径主要调节免疫系统功能[39]。非经典NF-κB激活主要表现为抑制性蛋白的降解:非激活状态下,TRAF3与TRAF2-cIAP1/2形成E3复合体,降解非经典NF-κB途径关键激酶——NF-κB诱导激酶(NF-κB-inducing kinase,NIK);信号激活时,TRAF3等抑制性蛋白被泛素化降解,NIK的抑制解除,NIK活化并向下传递细胞信号[37]。OTUD7B可通过其N-端UBA结构域结合TRAF3并催化其K48-脱泛素化,抑制TRAF3进入UPS降解途径[10]。OTUD7B基因敲除的小鼠中,B淋巴细胞发生抗原超应答,小肠黏膜淋巴滤泡增生,对肠道细菌Citrobacterrodentium的防御能力增强[10]。OTUD7B通过调节TRAF3稳定性参与机体免疫调节,有望成为针对黏膜免疫治疗和非经典NF-κB信号相关疾病治疗的新靶点。
4.3调节肿瘤血管新生 恶性实体肿瘤的生长过程面临着氧气和营养需求不断增长和供应不足的矛盾。当供氧不足时,HIF-1α亚基泛素化降解受到抑制,其细胞内蛋白水平迅速升高,启动新的转录调控程序上调各种促血管新生因子的表达,促进细胞存活,加速肿瘤新生血管形成[40-41]。OTUD7B可结合HIF-1α并促进其K11-脱泛素化,维持HIF-1α蛋白稳定性,影响 HIF-1α介导的基因转录[13]。尽管HIF-1α和HIF-2α在基因序列上有很多相似性,但在组织分布和功能特性上有着明显的差异[42]。OTUD7B并不能直接调节HIF-2α蛋白水平,而是通过调控其上游转录因子E2F1的脱泛素化,维持E2F1稳定性,间接调控HIF-2α基因转录[43]。
4.4T细胞免疫反应和T细胞功能的调节 T细胞在抗感染和抗肿瘤免疫反应中发挥重要作用。外界抗原通过T细胞受体(T-cell receptor,TCR)激活初始T细胞(naive T cells),启动T细胞分化[44]。在TCR与配体结合活化后,OTUD7B可迅速募集到TCR信号核心分子酪氨酸激酶Zap70上并使其发生脱泛素化,从而抑制Zap70泛素化,阻止负性调节的磷酸酶Sts1和Sts2与Zap70相互作用,促进Zap70磷酸化和TCR信号转导,从而促进T细胞活化和T细胞介导的免疫反应和自身免疫反应[45]。
5 总结
OTUD7B是一种重要的脱泛素酶,通过靶向K11-、K48-和K63-连接的多聚泛素链,特异性调控底物蛋白的泛素化水平,参与细胞增殖、炎症反应、肿瘤血管生成和抗肿瘤免疫等多种信号转导途径的调节(表1),在肺癌、乳腺癌和肝癌等恶性肿瘤发生发展中发挥重要作用。OTUD7B的表达模式和作用机制受到局部细胞和组织内环境的影响,深入认识OTUD7B调控细胞生物学行为的分子机理,寻找其影响恶性肿瘤进程的组织特异性机制,有助于揭示恶性肿瘤泛素化调节的内在规律,为开发靶向OTUD7B的抗肿瘤治疗药物提供有力的线索和依据。

表1 OTUD7B介导的肿瘤相关信号通路
猜你喜欢
生物化学与生物物理进展(2022年8期)2022-08-20
材料与冶金学报(2022年2期)2022-08-10
湖北农业科学(2022年11期)2022-07-18
河北农业大学学报(2022年2期)2022-04-26
心血管病学进展(2021年8期)2021-09-13
科学与财富(2021年33期)2021-05-10
湖北农业科学(2020年24期)2021-01-21
实用肿瘤学杂志(2020年4期)2020-12-08
作文成功之路·小学版(2020年6期)2020-07-27
作文成功之路·小学版(2020年5期)2020-06-11
