
兔皮胶原快速明胶化分子机制
2019-09-18 09:12苏现波马明思戴宏杰周鸿媛张宇昊
食品科学 2019年17期
冯 鑫,苏现波,马明思,马 良,戴宏杰,余 永,郭 婷,周鸿媛,张宇昊,*
(1.西南大学食品科学学院,重庆 400715;2.邯郸学院生命科学与工程学院,河北 邯郸 056005)
近年来,我国兔肉产业发展迅速,产量约占世界总产量的40%[1]。兔皮是兔肉加工副产物,也是制备明胶的优质资源。传统的猪、牛源明胶具有良好的凝胶特性,但因疫病和宗教等原因使用受到很大限制;水产明胶在宗教和疫病方面不受限制,但因其亚基氨基酸含量偏低,导致凝胶特性较差,无法被广泛应用。本课题组前期研究表明,兔皮明胶的凝胶特性、起泡性、乳化性、成膜性等功能特性均不逊于猪皮明胶[2-7],且没有疫病威胁,宗教限制程度也远低于传统的猪、牛源明胶。因此,具有良好的应用前景。
此外,兔皮用于制备明胶的最大优势在于兔皮可经稀酸处理后快速实现明胶化。Yu Wei等[8]以兔皮为原料提取明胶,结果表明质量分数为1%的盐酸处理10 min可制得较高得率和凝胶强度的兔皮明胶;Ma Mingsi等[9]对兔皮胶原快速明胶化的机理进行了初步研究,结果表明,在酸处理过程中,兔皮胶原纤维中的共价交联可在短时间内被破坏,进一步延长酸处理时间则会造成胶原中亚基组分的降解,使后续清洗过程中损失率提高,进而降低明胶的得率和凝胶特性。此外,随着酸处理的进行,兔皮胶原中氢键被破坏,并在酸处理1 h后形成新的平衡,此时明胶化已经完成。该研究初步明确了兔皮快速明胶化的机理,但在明胶化过程中兔皮胶原分子结构的变化规律方面并未得出明确结论。
分子模拟又称“计算机模拟”或“计算机实验”,是一种根据实际体系在计算机上进行的实验,通过比较模拟结果与实际体系的实验数据来检验模型的准确性,并可检验由模型导出的解析理论所作的简化近似是否成功[10-12]。目前分子模拟技术已发展成为生物信息学、分子生物学、化学和实验科学等领域中一种重要的研究工具[13-15]。通过分子模拟可建立多肽、蛋白质分子的初始模型,也可显示已被测定的蛋白质分子的空间结构,帮助和促进蛋白质肽链折叠和蛋白质结构的预测、总结蛋白质的结构规律、揭示蛋白质结构与功能的关系[16-20]。最初分子模拟的应用主要集中在分子生物学以及化学领域,在食品研究领域的应用较少,其主要原因在于多数食品蛋白并不像β-乳球蛋白具备完全解析的结构模型[21-23]。但随着蛋白质数据库的不断完善和同源建模预测技术的不断发展,使得以已知蛋白质结构为模板,通过对比蛋白的一级序列来筛选和预测未知蛋白质高级结构成为可能[24-26]。目前在蛋白数据库中,并无完全解析的胶原蛋白模型,但在数据库中能够检索到兔皮胶原较为完整的氨基酸序列,为胶原蛋白模型的构建提供了依据。
因此,本研究在实验室前期兔皮胶原快速明胶化的基础上,依据检索到的兔皮胶原一级序列,采用分子模拟技术构建相应的模型结构,依据实际实验调整设置酸处理环境,模拟兔皮明胶化过程,以期全面解析兔皮胶原明胶化过程中胶原的结构变化,为实现兔皮明胶的科学、高效制备提供理论参考。
1 材料与方法
1.1 模型构建
通过Uniprot(https://www.uniprot.org/)查找兔皮I型胶原α1链和α2链氨基酸序列,并以此作为建模的依据。
非螺旋区域:胶原蛋白的非螺旋区模板通过NCBI BLAST进行检索,选择E-value得分以及比对结构得分(total score)较优的蛋白结构作为模板结构。通过YASARA软件对胶原蛋白三维结构进行同源建模。
三螺旋区域:三螺旋结构具有典型性,可采用CCBuilder Mk.2软件直接进行模型构建。
1.2 模型评价
分别用PROCHECK和ERRAT程序对优化后模型蛋白结构进行评价。
1.3 单体系动力学模拟
以YASARA动力学软件对胶原蛋白进行动力学模拟,使用AMBER14力场,体系压力为1×106Pa、溶剂密度为0.997 g/L、pH值为1.02、模拟温度为25 ℃。I型胶原蛋白的三螺旋结构通过CC Builder Mk.2软件构建,非螺旋肽结构来自于同源建模结果。确定模拟体系的温度耦合采用“Berendsen thermostat”法,压力耦合采用“manometer”方法,参照压力为3×107Pa。采用标准立方盒包裹模型及其他分子,复合物置于盒子中心,盒子设置成周期循环,并添加抗衡离子(Na+和Cl-)固定蛋白,使用最陡下降法对溶剂进行能量优化,然后再用局部最陡下降法消除原子间的碰撞;随后限制蛋白主链,使用最陡下降法对溶剂进行能量优化,然后再用局部最陡下降法消除原子间的碰撞;每100 ps保存一个轨迹。
上述方式进行模拟体系中的远程范德华力作用距离取0.8 nm,经典相互作用使用球型截断半径“cut-off”方法计算。使用YASARA软件包的动力学脚本(md_run.mcr)进行动力学模拟。
1.4 数据处理与分析
数据分析处理采用YASARA和Origin 8.0软件。
2 结果与分析
2.1 模型构建
胶原蛋白具有三螺旋区域和两端非螺旋区域,对2 个区域分别进行建模,结果如图1所示。

图1 胶原蛋白三维结构示意图Fig. 1 Schematic three-dimensional structure of collagen
2.1.1 非螺旋区模型构建
通过NCBI BLAST对非螺旋区域模板进行检索,发现胶原非螺旋区具有5K31、4AE2、4AEJ 3 个模板。以E-value得分以及比对结构得分(total score)为依据,对3 种模板进行筛选。选择较优的蛋白结构作为模板结构,进行后续的计算。兔皮胶原α1、α2链的氨基酸序列和3 个模板的序列比对结果如图2所示,模板相关信息见表1。

图2 肽链α1、α2非螺旋部分残基与模板5K31、4AE2、4AEJ 的序列比对Fig. 2 Sequence alignment of α1, α2 non-helical residues with 5K31,4AE2 and 4AEJ templates

表1 同源性模板序列比对结果Table 1 Results of homologous template sequence alignment
通过图2的序列比对可知,兔皮胶原氨基酸序列与模板的覆盖率为71%~74%,序列匹配一致性为63%~91%,具有较高的一致性。表1显示的是3 种模板的得分情况,一般来说总体分值高于300或者E值小于1 e-5的晶体可以作为候选模板进行同源建模[27]。由表1可知,5K31、4AE2、4AEJ 3 个模板均满足以上条件。为此,通过YASARA软件对3 个模板进行混合建模以进一步筛选,在这一过程中只有模板5K31通过了模型迭代,因此确定5K31为最终模板。所构建的非螺旋区模型如图3所示。

图3 非螺旋区结构模型示意图Fig. 3 Schematic representation of the non-helical region model
2.1.2 三螺旋区模型构建
胶原的三螺旋结构具有典型性,通过CC Builder Mk.2软件可直接进行I型胶原蛋白三螺旋区域模型的构建,模型构建结果如图4所示。

图4 三螺旋结构模型示意图Fig. 4 Schematic representation of the triple-helix model
由图4可知,构建的三螺旋结构的α1与α2链均为左手螺旋(图4A),3 个螺旋再形成右手超螺旋结构,并且3 条α肽链将相互交错排列(图4B),这些结构特征都与胶原蛋白结构相符合,说明通过此方法构建的三螺旋结构在空间构象上合理。
2.2 模型评价
采用PROCHECK和ERRAT程序对优化后模型蛋白的结构进行评价。拉氏图(Ramachandran plot)主要用于指明蛋白质或肽中氨基酸残基的允许和不允许的构象。拉氏图主要分为3 个区域:允许区(红色区域)、最大允许区(黄色区域)和不允许区(空白区域),图中的黑点代表氨基酸二面角。三螺旋结构模型的拉氏图显示,模型中100%氨基酸二面角位于允许区(图5)。而非螺旋肽结构模型99.2%的氨基酸二面角在合理的范围之内,2 个模型均符合立体化学能量规则。

图5 三螺旋结构模型和非螺旋肽结构模型的拉氏图Fig. 5 Ramachandran plots of the triple-helix and non-helix models
2.3 单体系动力学模拟
实际明胶化过程是将胶原蛋白在质量分数为1% HCl溶液中进行5 min~24 h不等时间的酸处理。在分子模拟过程中,计算机处理时间与实际时间代表的含义不同,综合考虑计算机的处理能力,在模拟过程中设定总处理时间为200 ns,旨在观察二级结构的变化情况。
2.3.1 非螺旋肽结构的RMSD分析结果
均方根偏差(root mean square deviation,RMSD)可以近似显示出体系构象的相对变化。单个、多个非螺旋肽结构的RMSD分别按式(1)、(2)计算。


式中:Rmsdi是第i个构象的RMSD,表示第i个构象与参照构象的平均偏差;Ne是构象数;σR是Ne个构象的Rmsdi涨落;Rd描述了Ne个构象的平衡程度。
由图6可知,对同源建模得到的非螺旋肽进行了200 ns的动力学模拟分析。在质量分数为1% HCl条件下,单个非螺旋肽结构的RMSD波动较小,只达到5 Å左右,说明酸处理对单个非螺旋肽结构构象变化影响较小。为此,本研究又观察了多个非螺旋肽结构的RMSD变化,相比之下,2 个非螺旋肽结构的RMSD波动较大,在100 ns时达到30 Å左右。在0~100 ns期间,2 个非螺旋肽结构的RMSD呈现上升趋势;而在100 ns后,2 个非螺旋肽结构的RMSD呈下降趋势。在180~200 ns期间,2 个非螺旋肽结构的RMSD渐稳定。

图6 I型胶原蛋白非螺旋肽结构RMSD随模拟时间的变化趋势Fig. 6 Change in of RMSD of non-helical peptides over simulation time
通过2 种体系的RMSD对比分析,发现多个非螺旋肽结构的RMSD波动十分明显,为进一步分析多个非螺旋肽结构在实验条件下RMSD波动较大的原因,每50 ns提取动力学轨迹进行作图分析,非螺旋肽结构随模拟时间的变化结果如图7所示。


图7 非螺旋肽结构随模拟时间的变化Fig. 7 Changes in non-helix structures over simulation time
随着模拟时间推移,原本结合在一起的非螺旋肽部分逐渐分离。这是由于在强酸条件下,非螺旋肽能够被质子化,在0~200 ns期间,非螺旋肽间的相互排斥作用使得非螺旋肽间彼此分离。0~100 ns期间非螺旋肽逐渐分散,相互排斥作用也导致非螺旋肽结构波动增加,RMSD升高。100 ns后多个非螺旋肽基本分离。由于非螺旋肽结构的紧密结合起着稳定胶原蛋白网络稳定的重要作用,因此在强酸条件下,多个非螺旋肽结构的分离将导致胶原蛋白的三螺旋肽结构易于被酸破坏。
2.3.2 非螺旋肽的二级结构变化分析结果
为进一步探究非螺旋肽在质量分数为1%的HCl条件下二级结构的变化情况,对此条件下非螺旋肽二级结构相对含量变化情况进行分析,结果如图8所示。

图8 非螺旋区域二级结构含量随模拟时间的变化趋势Fig. 8 Changes in secondary structure contents in non-helical regions over simulation time
由图8可知,在酸处理条件下,相对于单个非螺旋肽,多个非螺旋肽的无规卷曲结构相对含量有所增加,β-转角结构相对含量有所下降。该结果说明,酸处理使得非螺旋肽的质子化,导致非螺旋肽间彼此相互排斥,原本紧密结合的β-转角结构在正电荷相互排斥过程中,部分向无规卷曲结构发生转换。
多个非螺旋肽的二级结构变化显示,α-螺旋结构相对含量稳定在15%左右,β-折叠相对含量为30%左右,β-转角和无规卷曲二者相对含量总和在50%~60%左右。从二级结构变化情况可以看出,多个非螺旋肽各个二级结构相对含量虽然有一定变化但是并不明显,在酸处理条件下,该部分结构彼此分离后,空间结构并不会有太大改变,说明该部分不是引起实验上二级结构数据变化的主要区域。
2.3.3 三螺旋结构的RMSD分析结果
为了观察三螺旋结构在酸处理条件下的构象变化,对单个三螺旋结构进行了200 ns的动力学模拟分析,RMSD的分析结果如图9所示。在0~30 ns期间,RMSD呈现上升趋势,在30 ns时RMSD波动达到30 Å。在30~50 ns期间,RMSD逐渐稳定,在27~30 Å范围内波动,系统评价50 ns后RMSD将一直处于平稳状态,说明50 ns后易于明胶化部分的三螺旋结构已基本被破坏,剩余组分结构较为稳定,在短时间内不会发生变化,这与兔皮胶原明胶化过程中实际酸处理1 h内的情况类似,同时也说明相比于非螺旋区域,三螺旋区域的变化较为迅速,这可能是因为非螺旋区对三螺旋结构的稳定具有保护作用,伴随着非螺旋肽的分开,原本结合在一起的胶原分子被分离,导致三螺旋结构充分暴露在酸环境中,其结构的稳定性更易被破坏。

图9 单个三螺旋结构的RMSD随模拟时间的变化趋势Fig. 9 Changes in RMSD in single triple-helix structure over simulation time
2.3.4 三螺旋结构氢键含量分析结果
生物体系中最普遍、最基础的物质蛋白质的结构和功能都与氢键密切相关[28-30],在结构上蛋白质最重要的二级结构是由氢键决定的。为保证模拟过程更接近真实情况,对多个三螺旋结构的氢键变化情况进行分析,结果如图10所示。
在0~50 ns,三螺旋结构的氢键数量从开始的225下降到175,说明此时被质子化的三螺旋结构由于正电荷排斥作用导致其结构稳定性降低,原始构象遭到了破坏;在50~200 ns期间,三螺旋结构的氢键数量基本保持在175左右,此时氢键数量处于相对平衡状态,这可能是因为肽链间相互折叠卷曲不断形成新的氢键。本课题组先前有研究表明:实际明胶化过程中酸处理5 min使胶原的原结构破坏,氢键数量降低,1 h后随酸处理时间延长,氢键数量维持稳定[9],故说明酸处理1 h与分子模拟50 ns的结果具有高度一致性,对于兔皮胶原来说,酸处理1 h以内为最有效明胶化时间。
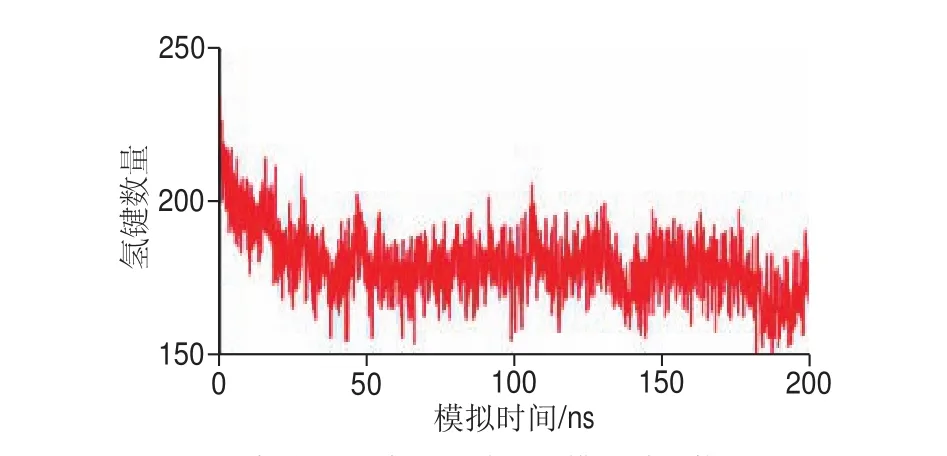
图10 三螺旋结构中氢键数量随模拟时间的变化情况Fig. 10 Changes in the number of hydrogen bonds in a three-helix structure over simulation time
2.3.5 三螺旋区的二级结构变化分析结果
三螺旋结构是胶原蛋白特有的二级结构,在酸处理中,三螺旋结构逐渐被破坏,胶原肽链逐步转化为其他二级结构。为进一步分析三螺旋区域二级结构的变化规律,对兔皮胶原最有效的明胶化时间即分子模拟50 ns(实际酸处理1 h)内,每10 ns提取动力学轨迹进行分析,分析结果如图11所示。

图11 三螺旋结构随模拟时间的变化Fig. 11 Changes of three-helix structures over simulation time
未处理时,三螺旋结构完整有序;当加入酸处理条件时,胶原三螺旋空间结构立即被打破,肽链松散呈无序状态,二级结构逐渐暴露。因软件无法区分松散的三螺旋结构和无规卷曲结构,在此本研究列出了其他3 种二级结构相对含量的变化趋势。随着酸处理时间的延长,3 种有序二级结构的总含量整体呈现增加趋势,说明三螺旋结构逐渐被破坏部分转化为其他二级结构。在3 种结构中α-螺旋和β-折叠相对含量始终未超过10%,原因在于胶原三螺旋区域含有大量脯氨酸(Pro)和羟脯氨酸(Hyp),这2 种氨基酸无法参与α-螺旋和β-折叠形成,但α-螺旋和β-折叠相对含量之和在明胶化过程中随着酸处理时间的延长而增加,到40 ns时达到最大值,而后开始下降,说明此时胶原中肽链中空间结构进一步展开,转化为无规卷曲,同时氢键达到平衡,展开的肽链在酸的作用下可能开始降解,造成得率开始下降。本课题组前期研究过程中发现α-螺旋和β-折叠相对含量之和与明胶得率具有较强相关性[9],与本研究结果一致,说明当α-螺旋和β-折叠相对含量之和达到最大时,明胶化程度最优。
综上所述,酸诱导兔皮明胶化过程中,随着酸处理开始,胶原非螺旋区亚基在酸的作用下开始分离,胶原三螺旋结构被破坏,转化为其他二级结构,当胶原肽链中α-螺旋和β-折叠相对含量之和达到最大值时,胶原明胶化程度达到最优。
3 结 论
对兔皮明胶化过程进行分子模拟,通过模板选择和YASARA同源建模构建出了兔皮胶原的非螺旋结构,采用CCBuilder Mk.2软件构建出兔皮胶原的三螺旋结构。采用PROCHECK和ERRAT程序对优化后模型蛋白结构进行评价,2 个模型均符合立体化学能量规则。Verify-3D检测结果表明,2 个模型质量良好。RMSD分析结果表明,随着酸处理的进行,蛋白分子的非螺旋区亚基分离,但其中单个非螺旋区构象变化较小。三螺旋结构的氢键数量变化表明,酸处理导致三螺旋结构的氢键数量在短时间内迅速减少,模拟后期氢键数量保持相对稳定,处于平衡状态,与实际酸处理1 h内的明胶化胶原红外光谱的结果一致。三螺旋结构随模拟时间的变化结果表明,随着非螺旋区亚基结构的分离,胶原三螺旋结构被破坏,转化为其他二级结构;α-螺旋和β-折叠相对含量之和呈现先上升后下降的趋势,当α-螺旋和β-折叠相对含量之和达到最大时,明胶化程度最优。
猜你喜欢
化工管理(2022年13期)2022-12-02
河北果树(2022年1期)2022-02-16
南昌大学学报(理科版)(2021年3期)2021-10-13
波谱学杂志(2021年3期)2021-09-07
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
华北农学报(2020年5期)2020-11-10
天然气工业(2019年10期)2019-11-12
天津师范大学学报(自然科学版)(2016年4期)2016-12-14
中学化学(2015年12期)2016-01-19
中国塑料(2014年9期)2014-10-17
