核壳结构纳米相析出的第一性原理界面热力学
2019-09-18 00:47:00张朝民
中国材料进展 2019年8期
江 勇,张朝民
(1.中南大学材料科学与工程学院,湖南 长沙 410083)(2.中南大学 粉末冶金国家重点实验室,湖南 长沙 410083)
1 前 言
析出强化(或称时效强化)合金的发现已有100多年的历史,已成为目前各个工业领域金属结构材料应用最广泛的强化机制之一[1, 2]。经热处理,在过饱和合金基体中析出大量细小、弥散的第二相颗粒,可以有效阻碍位错和晶界运动,提高合金强度。决定实际析出强化效果的因素很多,包括析出相尺寸、形貌、分布、共格度以及热稳定性等。近10年来,随着实验表征分析和计算机技术的飞速进步,人们已经可以在亚纳米尺度对合金析出相进行深入的微观结构表征和能量学计算。其中原子尺度表征技术包括高角度环形暗场像扫描透射电镜(HAADF-STEM)[3, 4]、高分辨透射电镜(HRTEM)[5, 6]、球差校正扫描透射电镜(Cs-corrected STEM)[4, 7]和三维原子探针层析技术(3D-APT)[8-13]等,而常用的计算方法包括分子动力学(MD)[14, 15]、动力学蒙特卡洛(KMC)方法模拟[16-18]以及基于密度泛函理论的第一性原理计算[19, 20]等。通过原子尺度上的表征和计算研究,人们能够更全面深刻地理解和认识合金析出相的形核热力学、长大动力学,尤其是其析出形貌、尺寸、分布、演变以及与合金宏观性能的相关性等。
多元析出强化型合金在时效过程中,过剩溶质原子可能偏聚到先析出的第二相界面,在降低界面能的同时,可以把第二相颗粒包裹起来,形成溶质富集的过渡原子层或新的界面析出相。这种类似核壳结构的形成,一般有利于提高先析出相的结构稳定性,有效抑制其长大以及后续发生的成分和结构演变,从而对合金微结构和力学性能的稳定性有较大提升。以铝合金为例,目前研究报道较多的具有核壳析出结构的铝合金是三元Al-Sc-Zr合金[9, 13, 17, 21-24]。实验发现,相比Al-Sc二元合金,三元Al-Sc-Zr合金经等温时效后可均匀弥散析出高数密度、高度共格、纳米级的L12-Al3(ScxZr1-x)相。这种析出相具有内核富Sc和外壳富Zr的典型核壳结构,可有效钉扎位错、晶界和亚晶界,提高合金强度、抗再结晶和抗蠕变性能(图1)。除Zr以外,在Al-Sc合金里添加一些其它过渡族金属元素,同样可以形成类似的核壳结构析出相[6, 9-11, 13, 17, 25-32]。对Al-Sc-Li合金的研究也有类似结果的报道[7, 33, 34]。事实上,在Al合金中形成核壳结构析出相,并不必须依赖Sc的添加。Al-Er-TM[5, 35-37]和Al-Yb-Zr[38-40]中同样可以形成核壳结构纳米析出相,对合金的力学性能、热稳定性和抗蠕变性都有所提升。在三元Al-Sc-Yb合金基础上进一步添加Li,经双级时效后甚至还可以析出双壳结构的L12相[41, 42],即在富Yb的内核之外依次形成富Sc和富Li的双壳层。但从析出强化机制上看,更复杂的双壳结构并不一定比单壳结构具有更优的强化效果[43]。

图1 合金中弥散细小析出相对位错和晶界运动的阻碍示意图。相对于单一结构,核壳结构析出相可能具有更高的热稳定性,有利于提高合金高温和蠕变强度Fig.1 Schematic of the motion of dislocations and grain boundaries hindered by dispersive fine precipitates in an alloy matrix. Compared with the uni-structured precipitates, the core-shelled precipitates may have higher thermal stability, in favor of the high temperature strength and creep strength of the alloy
核壳结构纳米相析出在其它合金体系中也有不少发现和报道。比如在某Fe-15Cr铁素体合金基体中析出的核壳结构纳米氧化物[44],其内核富Y-Zr-O,外壳富Ti,具体成分含量和结构受合金成分和制备工艺影响。这种核壳结构的形成,能够有效抑制纳米氧化物颗粒在高温(550~700 ℃)下发生粗化。原子数分数为9%~12%的Cr钢[45]中析出的核壳结构Z相,提高了合金的蠕变强度,其核壳结构的形成与Cr的扩散有关。另外,在Zn-Mn-Fe合金[46]中,MnZn13通过在先析出的(Fe, Mn)Zn13界面上外延生长,也形成了核壳结构。在Mg-Zn-Gd-Zr合金[47]中,也发现了内核为Zn2Zr相、外壳为Gd的核壳结构相的析出,其形成与富Zr的内核加速Gd的界面偏聚有关。经辐照后的Cu-W-Nb合金[16, 48],在退火过程中可析出内核富W、外壳富Nb的核壳纳米相,同样提高了析出相的抗粗化能力。在高熵合金Al2CrCuFeNi2[49, 50]中也发现了内核富Cu、外壳富α-NiAl的核壳纳米析出相。可以预计,近年来随着原子尺度实验表征技术的飞速发展,可能还会有更多类型的核壳结构析出相在更多不同合金体系中被发现和报道,与之同时,对核壳析出相成分、形貌、性质及其对材料宏观性能影响的研究方法和研究结果也将更加丰富。
目前有关复合结构析出相的理论研究,主要集中在对其形成热力学和动力学开展计算。例如运用相场计算模拟方法,借助界面能、应变能和扩散率等基础数据,可以预测Fe基合金中析出相的临界形核能垒、最低能量路径,进而预测析出相是否可能形成核壳结构[51]。基于Gibbs-Thomson效应,运用KMC方法可以计算评估三元合金中纳米析出相的抗粗化性能[16-18]。借助基于第一性原理的KMC计算模拟,可以分析Al-Sc-Zr合金中核壳结构析出相的形成,并将成因归结为Sc和Zr在Al基体中扩散速率的差异[17]。KMC计算模拟与3D-APT结合,可以深入分析Fe-Cu-Mn-Ni-Si合金中的共析出行为以及核壳析出相的结构和形貌[18]。与第一性原理计算相比,相场和KMC模拟更适合揭示析出相形成的动力学过程,但一般需要基于已有的实验结果来帮助确定一些经验性参数。虽然部分参数可以借助第一性原理直接计算获得,但由于自身无法精确考虑一些重要的量子效应,比如溶质原子的界面偏聚或析出相界面的应变能,常规的相场和KMC模拟方法往往只能给出定性而非定量的预测结果。
从热力学角度阐明纳米析出相及其界面的形成机制,定量计算预测其稳定结构和评估其稳定化趋势,第一性原理能量学计算为我们提供了更基础、更精确的方法。该方法通过建立体系的量子力学Schrödinger方程直接求解体系能量,进而通过建立体系能量与材料各种物理及化学性质之间的关系,推导出材料的各种物理及化学性质。对能量的计算结果依靠自洽的能量准则 (或原子间力准则)自我收敛,不需要依赖经验性参数或实验数据的输入,故可排除一切人为因素的影响,最终的计算结果可以做到自我支持。第一性原理计算在理解和预测溶质原子的固溶和偏聚,复杂纳米析出相及其界面结构、性质和形成机理方面,可以突破目前实验测试技术的诸多局限,已成为合金纳米析出相研究不可或缺的重要手段[52-54]。
本文以三元铝合金体系(Al-Sc-Zr和Al-Er-Zr)为例,采用第一性原理热力学计算研究方法,首先计算预测了Al/Al3Sc、Al/Al3Er的界面结构、界面能和应变能;进而计算预测了Al/Al3Sc和Al/Al3Er界面的Zr原子偏聚行为,包括偏聚结构和偏聚能;最后借助经典均匀形核理论,计算评估三元L12型析出相Al3(Sc, Zr)和Al3(Er, Zr)各种可能的析出结构及其热力学稳定性。基于以上计算研究结果,可以对文献中许多貌似冲突的实验观察给出合理解释。所涉及的研究方法并不限于Al合金,也可以广泛适用于其他析出强化型合金体系。
2 计算方法
本文所有的第一性原理计算采用基于密度泛函理论(DFT)的半商业化代码包-VASP(Vienna Ab-initio Simulation Package)[19, 20]。计算采用平面波基组和周期性边界条件,在冻芯近似下利用 Blöchl投影缀加波方法[55]描述离子实-电子相互作用。交换关联泛函采用Perdew-Burke-Ernzerhof (PBE)的广义梯度近似(GGA)[56]。平面波基展开的动能截断和布里渊区划分的K空间网格大小均进行了自洽的收敛性测试。L12结构析出相的体相计算采用常规单胞,收敛性测试表明20×20×20的Monkhorst-Pack K点网格和400 eV的能量截断足以满足体计算的准确性。所有界面结构的计算, Monkhorst-Pack K点网格密度设置尽可能接近体相的计算。所有电子步迭代的一致性收敛准则被设置为10-5eV/atom。所有结构的弛豫计算中,每个原子所受的Hellman-Feyman原子间力收敛于0.01 eV/Å以内。
3 Al-Sc-Zr合金中的核壳纳米相析出
已有高分辨电子显微镜的表征研究显示,Al-Sc-Zr合金中大量纳米尺度的核壳结构析出相具有与fcc-Al基体保持高度共格的L12型结构。纳米析出相计算涉及3种界面,即Al/Al3Sc(L12)、Al/Al3Zr(L12)和Al3Zr(L12)/Al3Sc(L12)。对于二元Al-Sc合金,主要强化析出相为L12-Al3Sc。添加少量溶质元素Zr,可能固溶于L12-Al3Sc相或偏聚到Al/Al3Sc(L12)界面,造成原有L12-Al3Sc相结构和性质的相应变化。探索Al-Sc-Zr合金中核壳结构纳米析出相的形成机理,可以首先从研究Al/Al3Sc(L12)界面开始。
3.1 Al/Al3Sc(L12)界面结构和界面能
异相界面原子结构模型的构建,必须考虑界面位向关系 (orientation relationship, OR)、实际接触面和界面终端结构、界面配位关系或堆垛方式以及相应的界面应变[57-61]。Al/Al3Sc (L12)界面的位向关系已被确定为(001)Al//(001)Al3Sc,[010]Al//[010]Al3Sc[62, 63]。考虑两者弹性模量的差异[64],对fcc-Al晶格仅施加~1.63%的拉伸应变与L12-Al3Sc相实现完全共格,这样构建的共格界面所需弹性应变能较低。计算选择了一组低指数界面作为实际接触面进行考察。Al3Sc{111}表面是典型符合化学计量比表面,只有一种Al3Sc表面终端结构,而Al3Sc{100}或{110}表面由Al和Al3Sc两种原子交替组成,可能有两种表面终端结构。界面超胞采用Al/Al3Sc/Al的三明治结构模型,其中Al块由5或7个原子层组成,Al3Sc块包含5到7个原子层,具体层数取决于界面接触面和界面终端类型。按照Al3Sc表面堆放fcc-Al晶格原子位置的不同,进一步考查了不同的界面原子配位类型,即顶位(top)、桥位(bridge)和孔位(hollow)。界面共格关系决定界面应变能,界面终端和原子配位关系决定界面原子之间化学成键,两者共同决定形成界面所需的能量,即界面能。
对界面超胞开展结构弛豫计算,可以确定(100)Al/(100)Al3Sc、(110)Al/ (110)Al3Sc和(111)Al/(111)Al3Sc界面的能量最低结构分别为Al终端+桥位、Al终端+孔位和Al3Sc终端+孔位(图2)。正视图中红色虚线为界面位置。俯视图中只显示了界面最近邻的上下两原子层,其中较大的实体球表示界面上层原子,较小的实体球表示界面下层的原子。
界面能衡量界面形成所需消耗的能量,可以计算表达为[34]:
ΔGf=GAl/Al3Sc-xNμAl-(1-x)NμAl3Sc
(1)
其中GAl/Al3Sc表示界面超胞的总能,N表示超胞中总原子数,x是其中Al的相分数。μAl或μAl3Sc是各相的标准化学势(即纯净单质相在其标准状态下的单位原子自由能)。这样计算的界面能实际包含以下两部分能量贡献,即克服两相之间晶格错配形成一定的共格关系而消耗的弹性应变能,和由界面结合形成新的界面化学键能。将以上能量平均到界面附近的每个原子,可以写成另一种表达方式[59, 65, 66],即:
(2)
A是超胞内所含界面的总面积,N是界面超胞中的总原子数。ΔGS是单位原子的界面应变能,γ则是不含应变的单位面积的界面能。对于给定的界面结构,按式(1)计算出的ΔGf/N与A/N数据画图,通过对式(2)的线性拟合,可以分别从截距和斜率中推导出界面应变能 (ΔGS) 和不包含应变的界面能 (γ)。由于式(1)中各相能量项之间存在大量相消,作为一阶近似,我们参照文献中的通用做法,暂且忽略热熵的贡献,以0 K基态下计算的焓变来评估界面形成能 (ΔGf)[34, 59, 66, 67]。
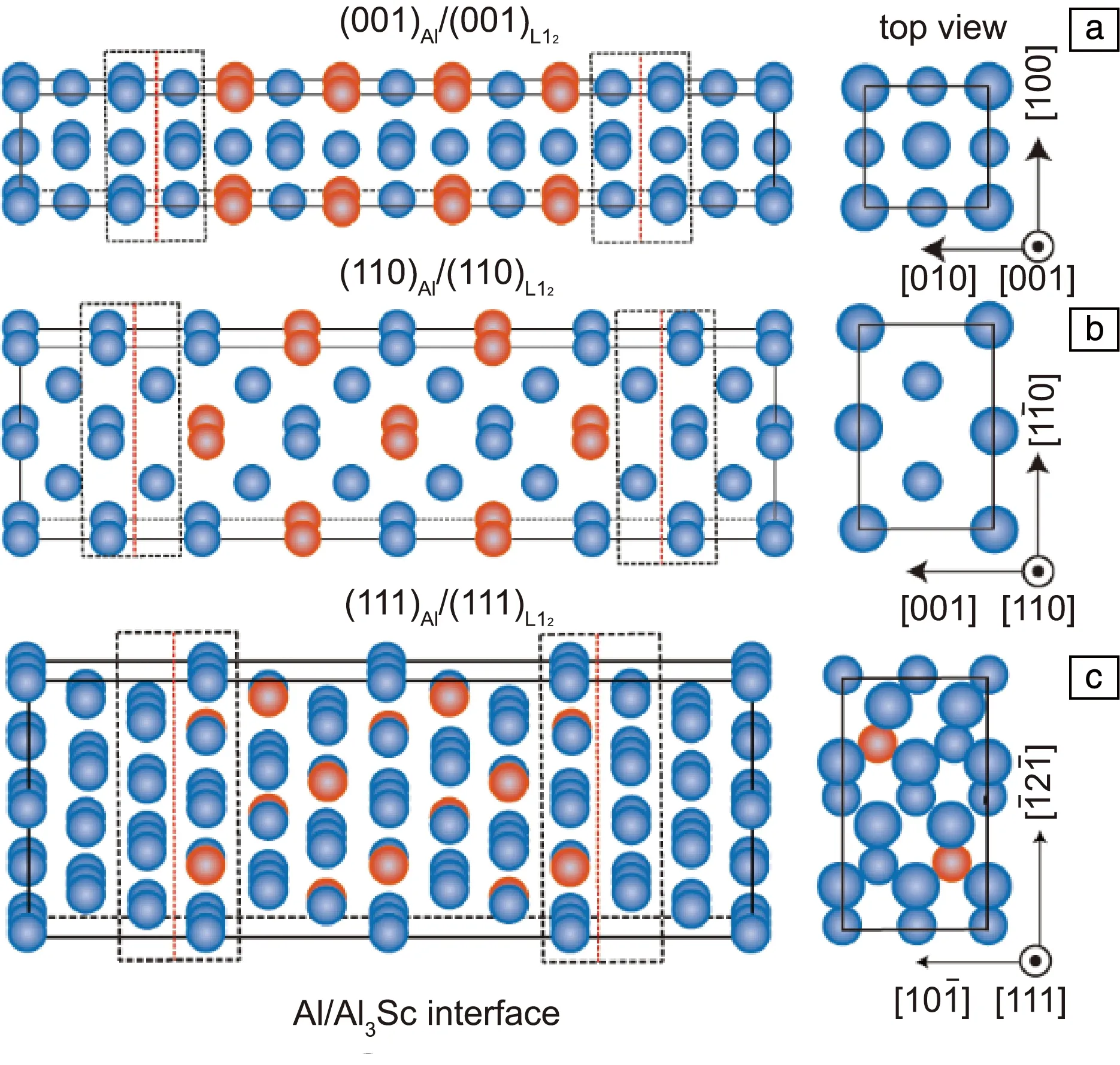
图2 计算用Al/Al3Sc(L12)界面超胞模型: (a) Al终端+桥位的(100)Al/(100)Al3Sc,(b) Al终端+孔位的(110)Al/(110)Al3Sc, (c) Al3Sc终端+孔位的(111)Al /(111)Al3ScFig.2 Al/Al3Sc(L12) interface supercells: (a) Al-terminated and bridge-coordinated (100)Al/(100)Al3Sc interface, (b) Al-terminated and hollow-coordinated (110)Al/(110)Al3Sc interface, (c) Al-terminated and hollow-coordinated (111)Al/(111)Al3Sc interface
从表1的计算结果可以看出,本文采用PAW-PBE赝势计算的界面能与文献中其他DFT方法计算的结果相对接近些,但均明显高于文献中基于嵌入原子势的分子动力学的计算值[70]。无论哪种计算方法,(100)Al/(100)Al3Sc界面能最低,对应的共格应变能也最低(表2),说明该界面是二元Al-Sc合金中析出L12-Al3Sc纳米相时最容易形成或热力学最稳定的界面。需要指出的是,表1中报道的实验值分别是基于析出相形核分析[72]和粗化分析[73]的推算得到的,数据结果比较分散,尤其是前者的推算结果过低。这可能是实验分析中考察的析出相较小,所含的原子数太少,没有严格定义的晶体相界面。相比于较大的析出相,低纳米尺度的团簇原子可以相对更自由地排列自己,从而最大限度地减少界面能[74]。由于以上实验研究方法均不能区分具体的界面原子结构,所报道的实测结果应视为是上述3种Al/Al3Sc(L12)界面能的一种平均。
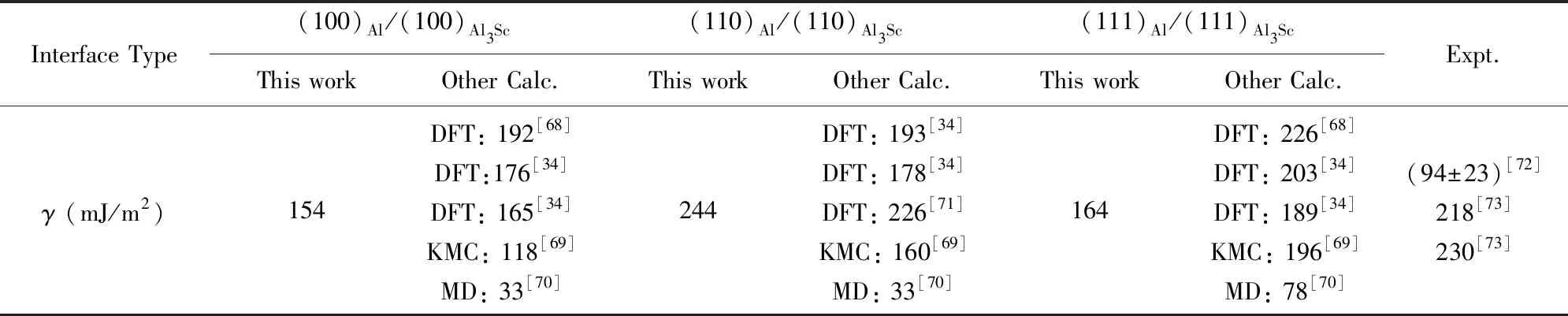
表1 计算得到的不包含应变的Al/Al3Sc(L12)界面能
3.2 Al/Al3Sc(L12)界面的Zr偏聚
为阐明核壳结构的形成机理,在界面计算的基础上进一步考察第二种溶质元素Zr对Al/Al3Sc(L12)界面的偏聚行为。如果Zr原子的界面偏聚具有热力学上的驱动力,偏聚到界面上的Zr就有可能取代界面上的Sc或Al原子,从而直接影响界面的化学成分、原子结构、相关能量和性质。
原子对界面偏聚的热力学驱动力一般通过计算偏聚能(ΔGseg)来评估,即
(3)
图3以(001)Al/(001)Al3Sc界面结构为例,显示了Zr偏聚到界面附近可能占据的不同原子层上的不同格点位。图4中计算比较了Zr偏聚到不同Al/Al3Sc(L12)界面不同原子层上的不同格点位所对应的偏聚能。结果显示:① Zr的界面偏聚行为强烈依赖于界面原子结构。② 无论哪种Al/Al3Sc(L12)界面,Zr总是倾向于偏聚到界面Al侧的第一原子层,并占据该层上的Sc格点位。相应的偏聚能计算结果见图4。这表明,基体中固溶的Zr原子偏聚到L12-Al3Sc先析出相界面的热力学驱动力明显。Zr具有较强的界面偏聚能力,表明了溶质原子Zr和Sc之间存在较强的相互作用。偏聚到界面的Zr原子与界面处最近邻的Sc原子处于fcc-Al晶格中的第二近邻(2NN)关系。已有计算证实[23],Zr与Sc原子处于2NN位置关系时存在一定的原子亲和力(~0.14 eV)。可以预见,大量过剩Zr原子的界面偏聚有可能导致形成一个包裹L12-Al3Sc先析出相的外壳,从而有助于抑制Al3Sc的粗化。③ 偏聚Zr不能取代L12-Al3Sc中的Al原子,但可能取代Sc原子,但对应的热力学驱动力(即偏聚能)很小。这种情况有可能在fcc-Al一侧第一原子层中的Sc位被偏聚的Zr原子占满后才出现。基于以上界面偏聚的定量计算结果可以推测,在Al-Sc-Zr合金中形成富Zr外壳结构的三元L12-Al3(Sc,Zr)相,不仅与扩散动力学有关,同时也可能具有强烈的热力学优势,因此也可能具备相应的结构稳定性优势。
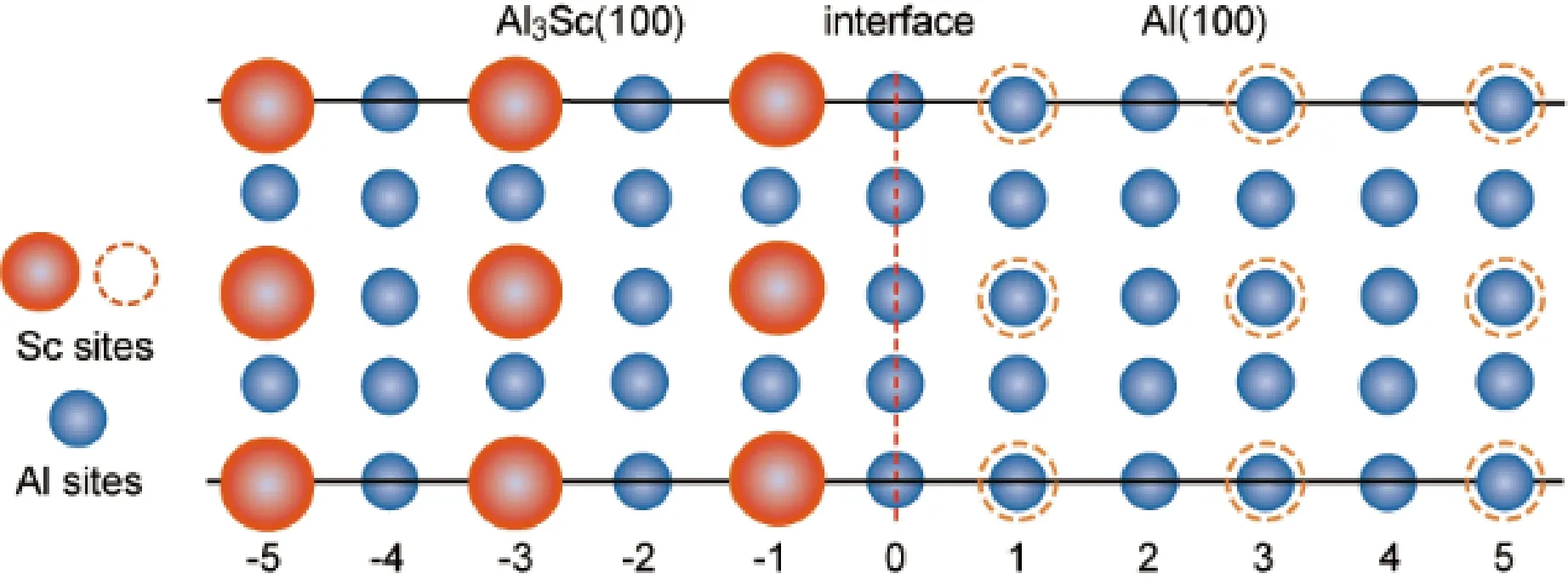
图3 (001)Al/(001)Al3Sc界面原子结构及偏聚Zr可能占据的格点位。蓝色球表示界面两侧各相中的Al格点位,桔红色球表示L12-Al3Sc相中的Sc格点位,桔红色虚线圆表示L12-Al3Sc相向fcc-Al相内部长大时将占据的Sc格点位。以上格点位均可能被偏聚的Zr取代Fig.3 Atomic interface structure of the (001)Al/(001)Al3Sc and the possible substitutional sites for segregated Zr. Blue balls denote the Al sites at both sides, and orange balls denote the Sc sites in L12-Al3Sc. Dashed orange circles denote the potential Sc sites in Al as L12-Al3Sc grows into the fcc-Al. All these type sites are possible to be occupied by segregated Zr

图4 对应不同Al/Al3Sc(L12)界面不同原子层上不同格点位计算得到的单个Zr原子偏聚能Fig.4 Calculated segregation energies for single Zr atom at different sites on different atomic layers of various Al/Al3-Sc(L12) interfaces
3.3 L12-Al3(Sc,Zr)纳米相的形核与结构稳定性
3.3.1 Al/Al3Zr(L12)和Al3Sc(L12)/Al3Zr(L12)的界面能
第3.2节中的计算已经证明了L12-Al3Sc为核的富Zr外壳可能是Zr原子偏聚到Al/Al3Sc(L12)界面后,在界面随机占位并富集的结果。一定条件下,这种富Zr外壳可能进一步演变,形成具有严格L12结构的Al3Zr相。为评估这种核壳析出结构的热力学稳定性,有必要将3.1节中的第一性原理界面计算扩展到Al/Al3Zr(L12)和Al3Sc(L12)/Al3Zr(L12)界面上。进一步借助经典形核理论分析,可以深入阐明复合结构纳米相的形核机制,并评估其析出结构的热力学稳定性。
文献调研发现,有关Al/Al3Zr(L12)和Al3Sc(L12)/Al3Zr(L12)界面的第一性原理计算目前还未有报道过。唯一针对Al/Al3Zr(L12)界面的理论计算基于动力学蒙特卡洛(KMC)模拟[69]。表3和表4总结的是采用3.1节中同样的方法,对以上两个界面开展第一性原理计算得到的结果。对比Al/Al3Sc(L12)界面的计算结果(见表1和表2),可以发现:① Al/Al3Zr(L12)界面和Al/Al3Sc(L12)界面一样,其(001)/(001)界面都是能量最低界面。② Al/Al3Zr(L12)的界面能普遍比Al/Al3Sc(L12)更低,表明在Al基体中形成该界面在热力学上更优。其中(110)Al/(110)Al3Zr界面能的第一性原理计算值为141 mJ/m2,与KMC计算值吻合较好。(001)Al/(001)Al3Zr和(111)Al/(111)Al3Zr界面能的第一性原理计算值明显低于KMC计算值,但似乎更接近实验推算的结果(100 mJ/m2)[75],后者同样应视为是所有可能的界面能的一种平均。③ Al3Sc(L12)/Al3Zr(L12)界面的界面能均为极小的负值,这么小的能量值已经接近总能计算收敛的误差标准,因此可以视为0,相应计算的界面应变能也接近0,说明该界面共格度非常高,界面结合强,尤其是热力学稳定性非常高,即该界面的形成可以不需要消耗系统总能,且一旦形成,能够保持清晰而稳定的相界。

表3 计算得到的Al/Al3Sc(L12)和Al3Sc(L12)/Al3Zr(L12)界面不包含应变的界面能(J/m2)

表4 计算得到的Al/Al3Zr(L12)和Al3Sc(L12)/Al3Zr(L12)界面的共格应变能
基于以上结果可以预测,在核相和壳相体积相等的条件下,L12-Al3(ScxZr1-x) 析出相的最佳策略应该是形成以Al3Sc(L12)核+Al3Zr(L12)壳的核壳结构(标记为L12-Al3Zr(Sc))。由于Al/Al3Zr(L12)和Al/Al3Sc(L12)界面能之间的差异,决定了该结构比Al3Zr(L12)核+Al3Sc(L12)壳的核壳结构(标记为L12-Al3Sc(Zr))更为稳定。核壳结构析出相一旦形成,能够在核壳之间保持稳定而清晰的Al3Sc (L12)/Al3Zr(L12)界面。实验[6, 13, 76]中分别观察到了各种不同形式的三元L12型纳米析出相,包括无序固溶的L12-Al3(ScxZr1-x)或具有不同核壳结构类型的L12-Al3Zr(Sc)和L12-Al3Sc(Zr),可能与Sc/Zr成分比例和析出量有关,也可能正处于向稳定核壳结构转变的某种亚稳状态。下一节中,将选择能量最低的(001)/(001)界面,基于经典的形核理论分析开展进一步的第一性原理计算研究,评估Al-Sc-Zr合金中可能形成的各种L12型纳米析出相的形成热力学和结构稳定性。提请注意的是,所有界面都基于基态的密度泛函第一性原理计算,这种近似一般会高估实际析出温度下的界面能[68, 69]。
3.3.2 析出相的形成热力学
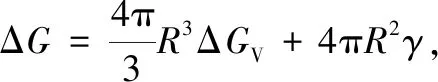
ΔGchem=G(Al3X)+(n-3)μAl-G(AlnX)
(4a)
或:
ΔGchem=[ΔH(Al3X)-ΔH(AlnX)]-T[ΔS(Al3X)-ΔS(AlnX)]
(4b)
公式(4b)中的焓变(ΔH)和熵变(ΔS)分别是L12-Al3X和fcc-AlnX(X=Sc或Zr)的形成焓和形成熵。提请注意的是,由于形核过程对温度很敏感,即便是一级近似的析出热力学计算,熵变的贡献也不应忽略。
计算得到的L12-Al3Sc相析出对应的焓差(ΔH(Al3Sc)-ΔH(AlnSc))为-0.776 eV/Sc,与已有实测值(-0.77 eV/Sc[77])和其它的DFT计算值(-0.72[34]和-0.76 eV/Sc[78])吻合很好。同样计算得到的L12-Al3Zr相析出对应的焓差是-0.831 eV/Zr。对于稀浓度合金来说,组态熵一般是可以忽略的,热电子熵在相对较低温度下也是可以忽略不计的[79, 80],故只考虑振动熵对ΔGchem的贡献。为了计算振动熵,在简谐近似下对L12-Al3X和fcc-AlnX(X=Sc或Zr)均采用3×3×3超胞模型计算了声子谱。计算得到的L12-Al3Sc相析出对应的振动熵差(ΔS(Al3Sc)-ΔS(AlnSc))为2.67 kB/Sc,与以前的DFT计算结果(2.66[34]和2.95 kB/Sc[80])接近。同样计算得到的L12-Al3Zr相析出对应的振动熵差为2.72 kB/Zr。当T= 673 K时,利用式(4)计算Al/Al3Sc(L12)和Al/Al3Zr(L12)的ΔGchem分别为-0.039和-0.050 eV/atom。已知Al/Al3Sc(L12)和Al/Al3Zr(L12)界面共格应变能分别为0.0038和0.0029 eV/atom(见表3),可以看出,从fcc-Al基体析出高度共格的L12-Al3Sc和Al3Zr相时,界面应变能与体积化学形成能在数值上相差约一个数量级。相应预测的临界形核半径分别为6.6和2.9 Å,临界形核功分别为~2.9×10-19和~2.9×10-20J。其中L12-Al3Sc的临界形核功(~2.9×10-19J)与文献中仅有的计算报道值(1.79×10-19J[81])接近,而L12-Al3Zr的临界形核功尚未见有文献报道。
以上计算显示,在fcc-Al基体中析出L12-Al3Zr和L12-Al3Sc,前者具有更小的临界形核半径和更低的临界形核功。换而言之,如果在某一给定温度下同时析出二元L12相Al3Zr和Al3Sc,在不考虑局域原子丰度的前提下,前者具有热力学上的优势,后者只具有动力学上的优势。
3.3.3 析出相的结构稳定性
上节中讨论和比较了L12-Al3Sc和Al3Zr相的形核热力学,其中L12-Al3Zr相的形核在热力学上优先,这也对应于第3.2.1节中计算预测的较低界面能。在稀浓度Al-Sc-Zr合金中,除了有少量L12-Al3Sc和Al3Zr的二元析出相形成,实验观察到了更多核壳结构[6, 13, 76]和非核壳结构[82]的L12型三元析出相。在本节中,选取在时效温度(T=400 ℃)和等溶质原子比(Sc和Zr原子比为1)条件下,计算评估三元L12析出相的各种可能结构的相对稳定性,包括L12-核壳结构 (Al3Sc核+Al3Zr壳结构,表示为L12-Al3Zr(Sc), Al3Zr核+Al3Sc壳结构,表示为L12-Al3Sc-(Zr))和无序均匀L12-结构 (表示为L12-Al3(ScxZr1-x)),并与二元析出相L12-Al3Zr和L12-Al3Sc进行比较。对相关结构稳定性的评估,我们基于经典形核理论,直接计算和比较各种结构的形核功如下[34]:
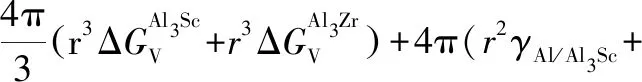
r2γAl/Al3Zr)
(5a)

4π(r2γAl3Zr/Al3Sc+R2γAl/Al3Zr)
(5b)
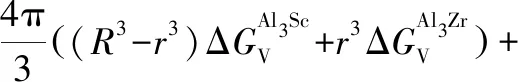
4π(r2γAl3Zr/Al3Sc+R2γAl/Al3Sc)
(5c)
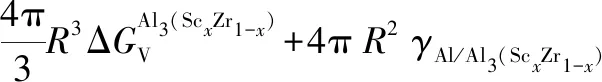
(5d)

图5为在T=673 K、Sc和Zr原子比为1条件下,计算得到的4种可能的析出相结构的总形成能与析出相半径的关系。其中黑色曲线为同时析出L12-Al3Zr和Al3Sc颗粒的计算结果,蓝色曲线为析出L12-Al3Sc(Zr)核壳结构,绿色曲线为析出无序均匀L12-Al3(ScxZr1-x)结构,红色曲线为析出L12-Al3Zr(Sc)核壳结构的计算结果。通过对比可以总结:① 各析出结构的相对稳定性为:核壳结构L12-Al3Zr(Sc)>无序均匀L12-Al3(ScxZr1-x)结构 > 核壳结构L12-Al3Sc(Zr)>L12-Al3Sc和Al3Zr的共同析出。② 在所考察的半径范围,各析出相结构的总形成能随半径的增大而增大。对于半径1~2 nm以内的析出颗粒,4种结构的形成能几乎相同,说明低纳米尺度下各种结构可能共存。③ 随着析出相半径不断增加,具有Al3Sc核+Al3Zr壳结构的L12-Al3Zr(Sc)相的形成能明显较低,由于其结构稳定性的相对优势明显,在稍大的析出相颗粒中将逐渐占居主导。计算所揭示的L12型纳米析出相尺寸与结构稳定性的热力学相关性,有利于澄清实验中HREM成像[6]和APT观察[9, 10, 13]结果的多样性。不同的实验表征对Al-Sc-Zr合金中L12析出相的研究结果不同,可能与合金溶质比有关,也可能是合金所处的不同热力学状态的直接反映。比如,对某种成分Al-Sc-Zr合金进行长达2412 h等温时效的实验后发现,随析出时间不断延长,L12析出相中富Sc核+富Zr壳的核壳结构的特征就越明显[9],这显然与计算预测的结构稳定性的变化规律完全符合,当合金体系不断接近其热力学平衡状态时,相对于其他可能的析出结构,核壳结构L12-Al3Zr(Sc)析出相的稳定性优势越发明显。

图5 在T=673 K、Sc和Zr原子比为1的条件下计算得到的4种L12型析出相结构的总形成能(或形核功)与析出半径的关系Fig.5 Predicted total formation energies (or nucleation energy) versus the precipitate radius for the four precipitation structures under T=673 K and the iso-atomic ratio of Sc to Zr is 1
4 Al-Er-Zr合金中的核壳结构纳米相析出
上节已经清晰地阐明了Sc和Zr的添加有利于形成具有Al3Sc核+Al3Zr壳结构的L12-Al3Zr(Sc)纳米析出相的热力学本质。由于Sc元素价格高昂,寻求替代元素获得类似高稳定性的L12型核壳结构纳米析出相,成为材料计算设计者的新动力。采用第3节中发展起来的计算方法,对Al-Er-Zr合金中可能析出复合结构L12-Al3-(Er, Zr)析出相进行了研究。具体计算细节不再赘述,这里只给出对应的计算结果。
4.1 Al/Al3Er(L12)界面及Zr偏聚
表5和表6分别为计算得到的Al/Al3Er(L12)界面的界面能和应变能。相比于上节中的Al/Al3Sc(L12)和Al/Al3Zr(L12)界面,Al/Al3Er(L12)界面能稍高,说明在同等热力学条件下析出,Al3Er可能更适合成为内核,可降低对体系能量的需求。同样地,在所有的低指数Al/Al3Er(L12)界面中,(001)Al/(001)Al3Er界面能量最低,这与其他Al/Al3X(X=Li, Sc, Zr)界面体系[34, 69]均类似。对Al/Al3Er(L12)界面能的计算预测值,均处于基于粗化实验模型推算出的(400±200) mJ/m2数值范围内[83]。

表5 计算得到的不包含应变的Al/Al3Er(L12)界面能(J/m2)
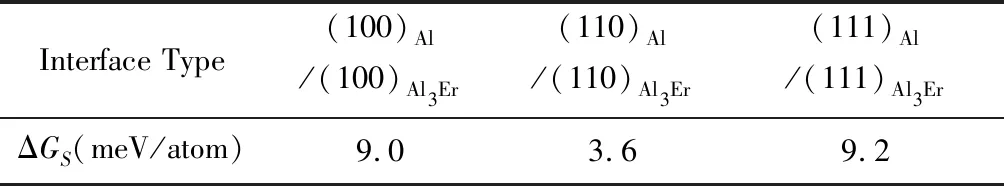
表6 计算得到的Al/Al3Er(L12)界面共格应变能
进一步计算预测Zr原子在Al/Al3Er(L12)界面附近不同原子层上的偏聚行为,结果见图6。和Al/Al3Sc(L12)界面类似,Zr对Al/Al3Er (L12)界面的偏聚行为同样强烈取决于受偏聚的界面原子结构,但无论哪种Al/Al3Er(L12)界面,Zr总是倾向于偏聚到界面Al侧的第一原子层,并占据该层的Er格点位。这表明Zr原子的界面偏聚同样有可能导致形成一个包裹L12-Al3Er先析出相的外壳,从而有助于抑制Al3Er的粗化。故可以推测,在Al-Er-Zr合金中形成核壳结构三元L12-Al3(Er,Zr)相,也可能具有热力学稳定性优势。
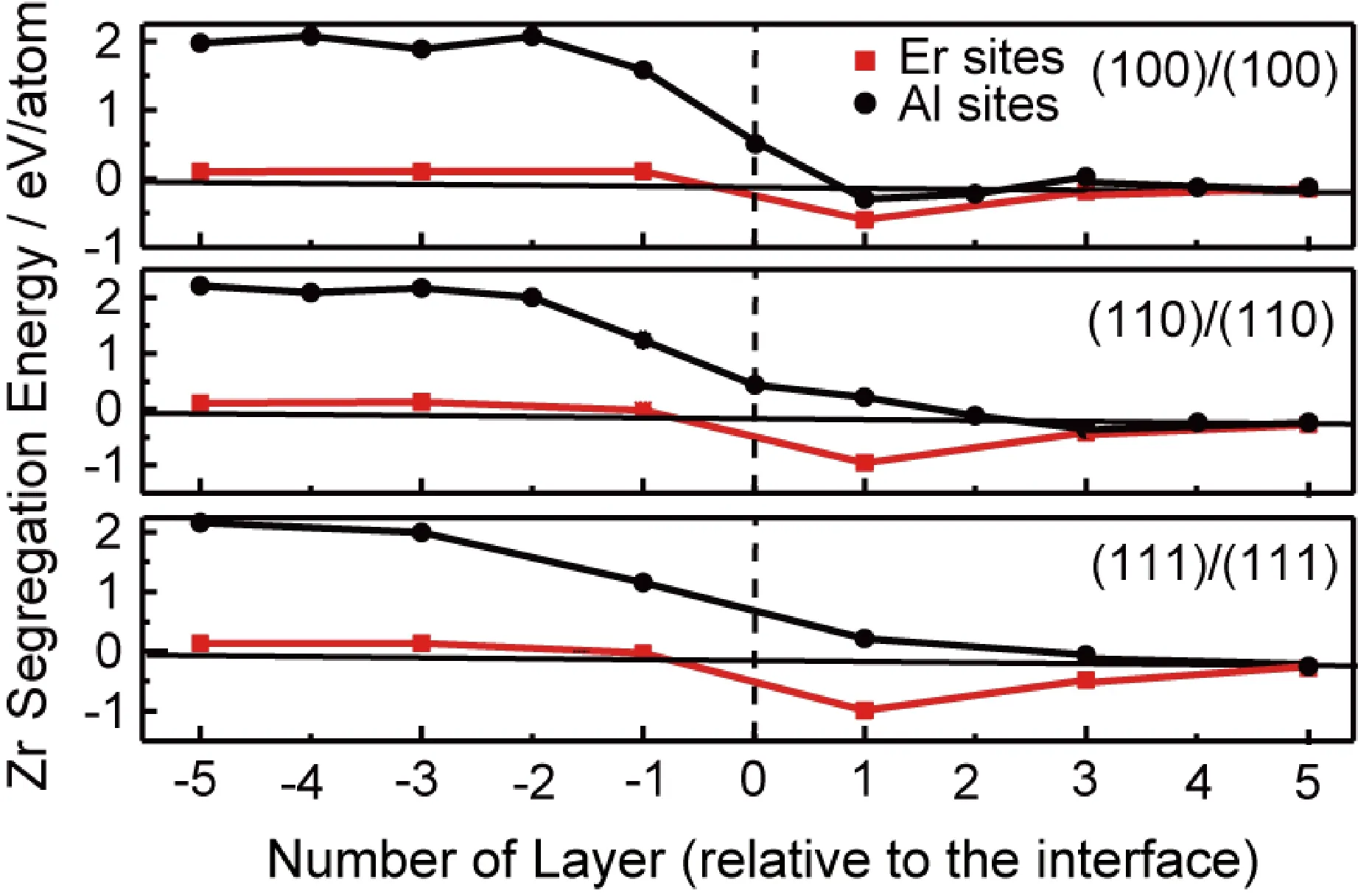
图6 对应不同Al/Al3Er(L12)界面不同原子层上不同格点位计算得到的单个Zr原子偏聚能Fig.6 Calculated segregation energies for single Zr atom at different sites on different atomic layers of various Al/Al3Er(L12) interfaces
4.2 L12纳米相的形成核与结构稳定性
将计算进一步扩展到Al3Er (L12)/Al3Zr(L12)界面,结合经典形核理论,深入分析和评估L12-Al3(Er,Zr)相的析出结构。针对该界面的计算研究,在文献中尚未见报道。
与Al3Sc(L12)/Al3Zr(L12)界面类似,计算预测的Al3Er(L12)/Al3Zr(L12)界面能也是非常小的负值,(100)/(100),(110)/(110)和(111)/(111)的界面能分别仅为-46,-47和-34 mJ/m2,在计算收敛的误差范围内,均可以视为0。其对应的共格应变能稍高,分别为5.0,5.1和4.4 meV/atom。显然,Al3Zr(L12)和Al3Er(L12)两相之间也同样容易形成稳定、清晰的界面。由于Al/Al3Zr界面能比Al/Al3Er更低,fcc-Al基体中析出的L12-Al3(Er, Zr)仍然可能是以较低界面能的Al3Zr为壳、Al3Er为核的核壳结构,且核壳之间能够保持一个清晰的相界面。
借用文献报道过的DFT计算值[84],按公式(4b)可以计算焓差(ΔH(Al3Er)-ΔH(AlnEr))为-0.867 eV/Er,对应的振动熵差(ΔS(Al3Er)-ΔS(AlnEr))为3.528 kB/Er。已知Al/Al3Er(L12)的共格应变能分别为0.009 eV/atom(表6),可以推算T=673 K下,Al3Er(L12)在Al基体中的临界形核半径为0.84 nm(与文献中其他DFT计算值0.87 nm[81]接近)。相应预测的临界形核功为~5.4×10-19J,与其他DFT计算值3.52×10-19J[81]接近。可见,在同样的条件下,L12-Al3Zr仍然比L12-Al3Er具有更小的临界半径和临界形核功,也更容易形核。
类似地,计算析出相的总形成能,以此比较4种结构的相对稳定性,即L12-核壳结构(Al3Er核+Al3Zr壳结构,表示为L12-Al3Zr(Er), Al3Zr核+Al3Er壳结构,表示为L12-Al3Er(Zr))和无序均匀L12-Al3(ErxZr1-x),并与二元析出相L12-Al3Zr和L12-Al3Er进行比较。图7为在T=673 K,Sc和Zr原子比为1的条件下,计算得到的4种可能L12析出结构的总形成能与析出相颗粒半径的关系。与Al-Sc-Zr体系类似,Al-Er-Zr合金中L12析出结构的相对稳定性为:核壳结构L12-Al3Zr(Er)>均匀L12-Al3(ErxZr1-x)结构>核壳结构L12-Al3Er(Zr)>L12-Al3Er和Al3Zr的共同析出。对于半径1~2 nm以下的析出颗粒,4种结构的形成能几乎相同而因此可能共存。随着析出相半径不断增加,具有Al3Er核+Al3Zr壳结构的L12-Al3Zr(Er)颗粒明显占优,形成能逐渐为负,表明这种核壳颗粒具有明显的结构稳定性优势,在所有L12型析出相中占绝对主导,与实验观察的结果吻合[5, 85]。其它结构的L12-Al3(ErxZr1-x)析出相只能是一种亚稳态,延长时效时间有助于接近和达到平衡,最终形成L12-Al3Zr(Er)的核壳结构。

图7 在T=673 K,Sc和Zr原子比为的1条件下计算得到的4种L12型析出相结构的总形成能(或形核功)与析出半径的关系Fig.7 Predicted total formation energies (or nucleation energy) versus the precipitate radius for the four precipitation structures under T=673 K and the iso-atomic ratio of Sc to Zr is 1
5 结 论
以三元铝合金为例,介绍了第一性原理热力学计算结合经典形核理论分析,应用于纳米共格核壳结构相的析出热力学和相对稳定性的研究实践。针对Al-Sc、Al-Zr和Al-Er二元合金体系的计算结果显示:① L12析出相与Al基体之间界面能高低次序是Al/Al3Er(L12)>Al/Al3Sc(L12)>Al/Al3Zr(L12),共格应变能高低次序是Al/Al3Er(L12)>Al/Al3Zr(L12)>Al/Al3Sc(L12)。② Zr在Al/Al3Sc(L12)和Al/Al3Er(L12)界面上均容易发生偏聚,并占据界面Al基体一侧的Sc或Er格点位,说明Zr的界面偏聚有利于核壳结构的形成。针对Al-Sc-Zr和Al-Er-Zr三元合金体系的计算结果显示:① 当析出相半径小于1~2 nm时,各种析出相结构均有可能共存,对应的形核能相差很小。当析出相半径大于1~2 nm,具有Al3Sc核或Al3Er核+Al3Zr壳结构的L12-Al3Zr(Sc)和L12-Al3Zr(Er)颗粒分别在两个合金体系中明显占优,表明这类核壳结构具有明显的热力学稳定性优势。② 由于二元L12析出相之间所形成的Al3Sc(L12)/Al3Zr(L12)和Al3Er(L12)/Al3Zr(L12)界面的界面能都极小,均接近0,表明核壳结构一旦形成,能够稳定维持清晰的核壳界面。③ 实验观察到的Al-Sc-Zr和Al-Er-Zr三元合金中的L12-Al3Zr、Al3Er和Al3Sc二元析出相,以及无序均匀的L12-Al3(Scx-Zr1-x)或L12-Al3(ErxZr1-x)三元析出相,均可能是正处于向稳态转变的亚稳态结构。
本文计算研究发现在给定温度和溶质比下,铝合金中共格L12相的析出结构及其相对稳定性与其尺寸有关,这有助于理解实验观测的多样性结果。而针对界面的第一性原理热力学计算研究方法,可以推广到其他合金体系中复合结构纳米析出相的研究,作为现有实验表征手段的重要补充,指导合金成分的科学设计。
猜你喜欢
上海金属(2022年4期)2022-08-03 09:52:10
原子与分子物理学报(2021年2期)2021-03-29 07:30:58
原子与分子物理学报(2021年1期)2021-03-29 07:29:40
原子与分子物理学报(2021年1期)2021-03-29 07:28:18
精密成形工程(2018年6期)2018-11-23 08:31:08
材料工程(2017年7期)2017-07-25 11:20:11
西安工程大学学报(2016年6期)2017-01-15 14:08:22
材料科学与工程学报(2016年1期)2017-01-15 13:34:08
贵州师范学院学报(2016年3期)2016-12-01 03:53:53
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10 08:41:29