
采用全自动在线固相萃取-高效液相色谱法检测畜禽产品中氟喹诺酮类药物残留量
2019-09-12 09:07:56程传民达列亚王宇萍樊淑娜
中国饲料 2019年15期
程传民,达列亚,李 云,柏 凡,魏 敏,王宇萍,李 茂,樊淑娜
(1.四川省饲料工作总站,四川成都610041;2.新疆维吾尔族自治区兽药饲料监察所,新疆乌鲁木齐833000)
氟喹诺酮属于促旋酶抑制剂四个亚型中的一种,一般将氟喹诺酮称为二代喹诺酮。其常作为兽用药物,被广泛应用于食用动物。氟喹诺酮是广谱抗生素,对多种细菌有抑制作用,特别是针对牛、猪和鸡。由于此类药物的大量使用,致使动物产生抗药反应,欧盟委员会制定的法令(EEC)No 2377/90,规定了一些氟喹诺酮类药物的最大残留限量(MRLs)。因此建立一种多组分、经济、方便、灵敏度高的全自动在线固相萃取-高效液相色谱检测方法很有必要。
1 材料与方法
1.1 仪器与材料
1.1.1 仪器 在线固相萃取-高效液相色谱仪(美国Aglient 1260);电子天平(0.01 mg,德国赛多利斯公司);涡旋振荡器(禾本森科有限公司);电动振荡器(美国IKA公司);高速冷冻离心机(Biofug PrimoR公司)。
1.1.2 材料 乙腈、甲醇(色谱纯,Fisher Scientific公司);乙酸(色谱纯,Fluka);0.1%三氟乙酸(用三乙胺调pH=3.0)(色谱纯,CNW);氟喹诺酮标准品(DR公司)。
标准品的配制:分别称取0.01 g(精确至0.1 mg)标准品置于10 mL棕色容量瓶中,用甲醇溶解,并定容至刻度,标准储备液浓度为1 mg/mL。将各标准储备液用20%乙腈溶液稀释,配成混合标准工作液,各组分浓度为10 mg/L,用20%乙腈溶液配制成目标药物浓度为1、2、5、10、50、100μg/L的混合标准系列溶液。
1.2 样品处理 称取均质畜禽产品样品2.00 g(精确至0.01 g)于50 mL离心管中,加入5 mL 1%乙酸乙腈(肉类加1 mL水,混匀后加4 mL 1%乙酸乙腈),涡旋提取2 min,在温度为4℃的条件下,10000 r/min离心5 min,上清液转入另一50 mL离心管中,重复提取一次。离心后合并上清液,加入20 mL正己烷,涡旋混合1 min,在温度为4℃的条件下,10000 r/min离心2 min,移去正己烷,取1 mL乙腈层到10 mL离心管中,加入4 mL水,过0.22μm滤膜后上机测定。
1.3 色谱条件 色谱柱:Aglient Eclipse XDBC18,规格150 mm×4.6 mm,粒径5μm,富集柱:C18的预柱。分析泵流速:1.0 mL/min;进样体积100μL;柱温为30℃。检测波长:激发波长280 nm;发射波长450 nm。分析泵流动相:A为0.1%三氟乙酸溶液(pH=3.0),B为乙腈溶液,流动相比例为A+B=88+12。运行时间35 min。上样泵流动相采用甲醇-水的二元溶液。采用溶剂梯度和流速梯度相结合的方式,具体见表1。阀切换:0~1 min:阀处于位置1,分析泵直接与分析柱联通,平衡色谱柱,上样泵将待处理的样品从自动进样器推到富集柱上进行富集和净化;1~2 min:阀处于位置2,分析泵与富集柱联通,将富集在柱头的待测样品反着冲到分析柱上进行分离分析,富集泵直接排废液;2 min以后:阀处于位置1,分析泵继续完成分离分析,富集泵与富集柱相连,冲洗并平衡富集柱,以备下一次上样。
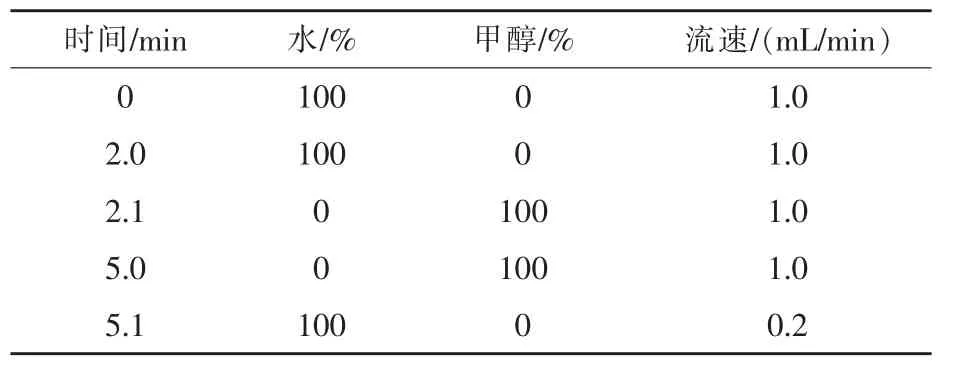
表1 上样泵液相色谱梯度洗脱条件
2 结果与讨论
2.1 流动相的选择 本实验研究了甲醇-乙腈-0.1%三氟乙酸和乙腈-0.1%三氟乙酸两种流动相对6种氟喹诺酮类药物分离度的影响,以甲醇-乙腈-0.1%三氟乙酸(5+8+87)为流动相时可以实现氟喹诺酮的基线分离,同时也可以多检测2种氟喹诺酮药物,增加的药物为氧氟沙星和沙拉沙星,当需要检测这两种氟喹诺酮时可采用此条件,但运行时间相当长,见图1a。以乙腈-0.1%三氟乙酸(12+88)为流动相时可以实现6种氟喹诺酮药物的分离,但诺氟沙星和氧氟沙星是完全没有分开的,由于氧氟沙星灵敏度相对较低,本研究选择做诺氟沙星,在样品检测时应注意药物的定性,色谱分离情况见图1b。
2.2 色谱柱的选择 本研究比较了Agilent poroshell 120 EC-C18 4.6 mm×100 mm,2.7μm、Agilent SB-C18 2.1 mm×100 mm,1.8μm和Aglient Eclipse XDB-C18,150 mm×4.6 mm,5μm色 谱柱,使用标准工作液对各规格色谱柱性能进行考察。Agilent poroshell 120在流速为0.4 mL/min的情况下,各目标化合物可以达到基线分离,但运行时间要到50 min。Agilent SB-C18在流速为0.3 mL/min的情况下,各目标化合物分离度不是很理想,色谱峰的对称性也相对差一点,可能和本实验使用的色谱柱的柱效有较大关系。若使用上述两种色谱柱还需进一步改变色谱条件。Aglient Eclipse XDB-C18色谱柱峰型和分离度可以满足本研究方法的要求,且一般实验室比较常见。
2.3 线性关系和检出限 将6种氟喹诺酮药物分别配制成质量浓度为1~100μg/L的系列标准溶液。采用本实验条件对系列标准溶液进行测定,以浓度X对所测物质的峰面积Y作图,得到线性方程以及线性相关系数。以实际可以检测出最低添加量为检出限。各目标化合物线性方程和检出限见表2。
2.4 样品测定 本实验对2个鸡蛋和2个鸡肉阳性样品进行检测,此检测结果与应用标准方法—鸡蛋和鸡肉中氟喹诺酮类药物及金刚烷胺残留测定操作细则(2018年全国例行检测使用的液质内标定量方法)的检测结果相对偏差小于8.0%,各目标化合物无杂质峰干扰,净化效果好,样品色谱图见图2。由此可见本实验方法与标准方法有较好的吻合,具体检测结果见表3。
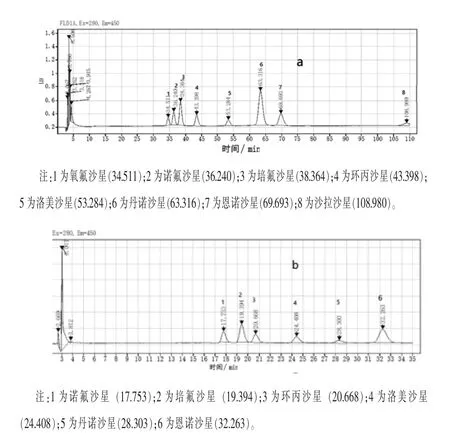
图1 氟喹诺酮类药物标准色谱图
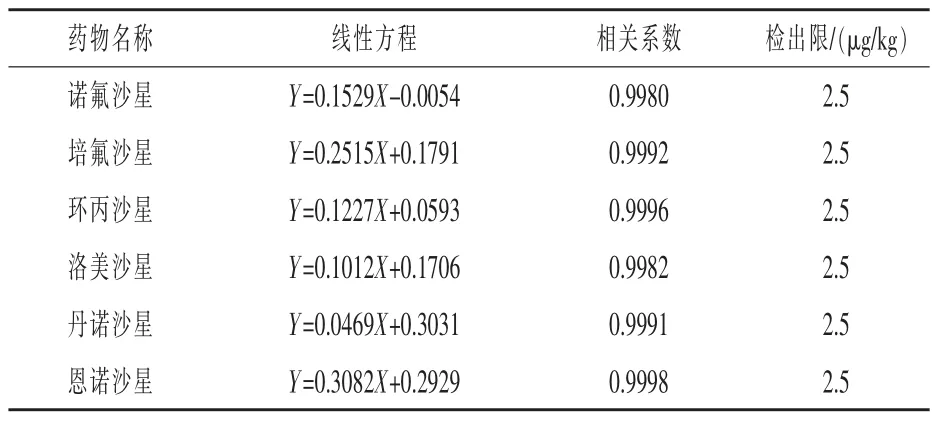
表2 6种氟喹诺酮药物的线性关系和检出限
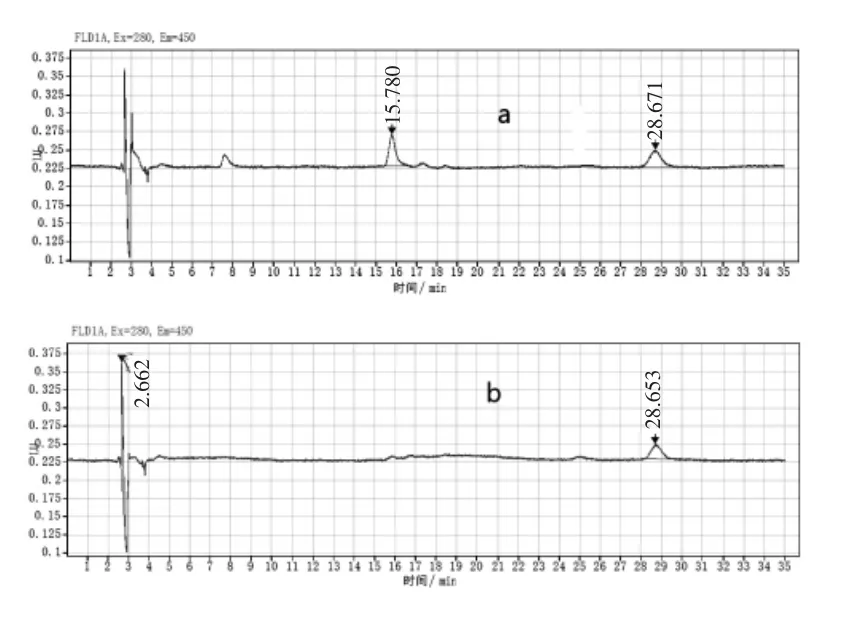
图2 阳性样品色谱图(a:鸡肉,b:鸡蛋)
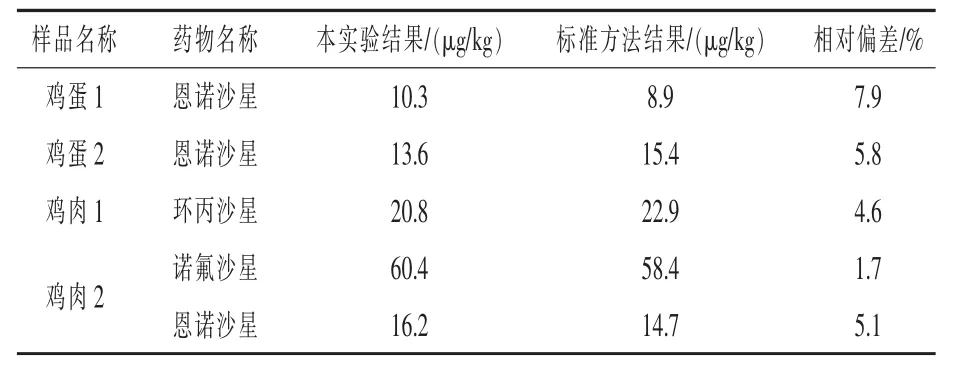
表3 阳性样品检测结果与使用标准方法检测结果对照
2.5 回收率和精密度实验 在3种不同类型空白试样中分别添加低水平和高水平2种剂量的氟喹诺酮药物。按照本实验方法进行加标回收实验,结果见表4。样品加标回收率(AR)为81.9%~106.8%。相对标准偏差(RSD)小于3.52%。该方法的回收率和重现性均较好。
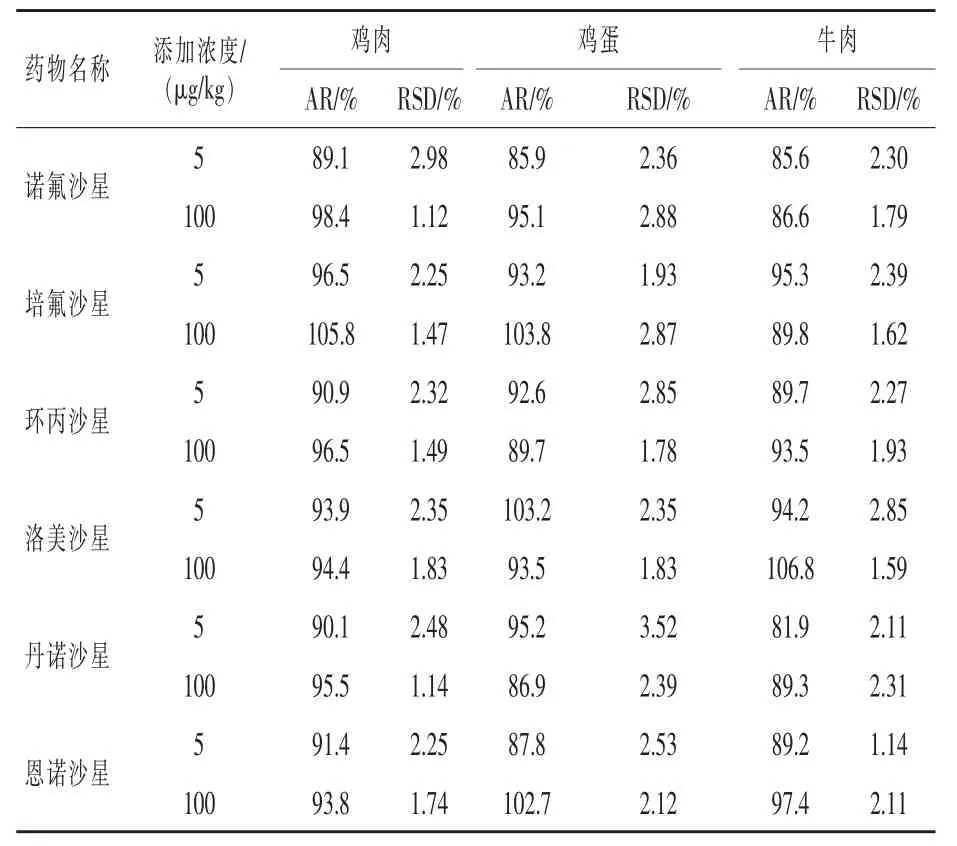
表4 回收率实验结果(n=6)
3 结论
本实验建立了同时检测6种氟喹诺酮药物的全自动在线固相萃取-高效液相色谱法,无干扰杂质峰,净化效果较好。6种氟喹诺酮药物在35 min内实现了基线分离,其线性相关系数大于0.9980,6种氟喹诺酮平均回收率为81.9%~106.8%;相对标准偏差小于3.52%;定量限为2.5μg/kg。该方法操作简单、经济、灵敏度高,结果准确可靠。
猜你喜欢
煤化工(2022年3期)2022-07-08 07:24:42
世界最新医学信息文摘(2021年12期)2021-06-09 08:36:56
浙江化工(2017年4期)2017-05-11 02:37:40
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:37
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
中国资源综合利用(2016年10期)2016-01-22 08:36:09
中国卫生标准管理(2015年25期)2016-01-14 09:29:24
含能材料(2015年1期)2015-05-10 00:50:52
中国实用医药(2013年36期)2013-09-11 01:13:06
天津化工(2010年5期)2010-09-18 02:55:58
