
双孢蘑菇6个菌株不同发育阶段的转录组分析
2019-09-10 07:22:44施肖堃卢园萍蔡志欣郭仲杰陈美元廖剑华
福建农业学报 2019年7期
施肖堃 卢园萍 蔡志欣 郭仲杰 陈美元 廖剑华

摘 要:【目的】通过多个双孢蘑菇菌株不同发育阶段的差异转录组分析,为进一步验证双孢蘑菇发育相关基因及探讨其分子机理奠定基础。【方法】对双孢蘑菇主栽品种As2796及其亲本02、8213,其回交子代W192,以及国外野生菌株ARP159、国内野生菌株AgLH830共6个具有重要代表性的菌株子实体原基期、幼菇期、采摘期、开伞期等4个不同发育阶段共24个样品进行转录组测序,并与双孢蘑菇参考基因组序列进行比对,根据比对结果进行各基因在不同样品中的表达量分析及差异表达基因识别,发掘新基因与共同基因的差异表达,并进行各数据库的基因功能注释。【结果】结果共鉴定到10 660个转录本,发掘新基因677个,其中237个得到功能注释。与原基期相比,6个菌株在幼菇期、采摘期和开伞期分别有49、82、73个共同差异表达基因,其中有13个是相同的基因。【结论】发现了一批在双孢蘑菇子实体不同发育阶段具有显著差异表达的基因,筛选出不同菌株不同阶段的共有差异基因,对双孢蘑菇子实体发育中重要的差异基因进行了注释与探讨。
关键词:双孢蘑菇;子实体发育;转录组测序;差异表达基因
中图分类号:S 646.11文献标识码:A文章编号:1008-0384(2019)07-775-07
Abstract: 【Objective】Transcriptome analysis was conducted on Agaricus bisporus at 4 developmental stages to identify the genes associated with and decipher the molecular mechanism involving the fungal development. 【Method】From 6 representative strains of A. bisporus including main cultivar As2796, its parents 02 and 8213, its backcrossing offspring W192, foreign wild strain ARP159, and domestic wild strain AgLH830, at primordium, young, harvesting, and opening stages, 24 fruiting body specimens were collected for transcriptome sequence analysis. By aligning them against the reference genome sequence of A. bisporus, the genes that were differentially expressed were identified. Both unique and common differentially expressed genes (DEGs) were clearly exposed to be annotated using the databases to determine their specific functions. 【Result】 Among the 10 660 transcripts obtained, 677 genes were unique with 237 functionally annotated. The tested A. bisporus shared 49 common DEGs between primordium and young stages, 82 between primordium and harvest stages, and 73 between primordium and pileus opening stages. And, 13 genes were found commonly present in the various strains. 【Conclusion】 Both unique and common DEGs in A. bisporus at the 4 developmental stages were identified and annotated in the study.
Key words: Agaricus bisporus; fruiting body development; transcriptome sequencing; differentially expressed gene
0 引言
【研究意義】转录组研究是发掘功能基因的重要途径,通过样本间基因表达差异比较,进而鉴定与特殊性状相关的候选基因,然后通过生物信息学方法和后续的实验对候选基因进行功能分析和验证,有利于在动植物疾病、功能基因组、育种等领域取得重要的科学发现[1-2]。转录组测序(RNA-Seq)是一种高效、快捷的转录组研究手段,通过高通量测序技术对样品中mRNA反转录形成的cDNA进行测序,统计相关读段(Reads)可计算出不同基因的表达量,该技术已被广泛应用于生物学、医学等研究领域。【前人研究进展】转录组测序在食用菌研究中也有应用,例如杨芳等[3]通过对鸡枞菌的转录组进行测序,分析发现了可能参与降解纤维素和木质素等相关酶类编码基因;Chen等[4]结合香菇基因组和转录组数据分析香菇木质素降解相关基因和转录因子的表达情况,揭示了该菌降解木质纤维素的遗传物质基础。Fu等[5]通过比较白灵菇不同发育时期的转录组数据,发现了一系列与子实体形成相关的基因。
【本研究切入点】前期对双孢蘑菇子实体不同发育与后熟时期开展了系列差异蛋白质组分析[6-9],在此基础上,本研究拟对双孢蘑菇Agaricus bisporus (J.E.Lange) Imbach国内主栽品种As2796及其亲本02、8213,回交子代W192,以及国外野生菌株ARP159,国内野生菌株AgLH830共6个具有重要代表性的菌株子实体原基期、幼菇期、采摘期、开伞期[8]4个不同发育阶段的样品进行转录组测序分析。【拟解决的关键问题】通过同一菌株不同发育阶段及不同菌株同一发育阶段间的差异比较,挖掘高产、优质等特性相关的基因,分析国内外菌株的异同点,发现与双孢蘑菇子实体生长、发育相关基因与通路,用于指导育种工作。
1 材料与方法
1.1 供试菌株
供试菌株As2796、02、8213、W192、ARP159、AgLH830由福建省农业科学院食用菌研究所保藏并提供。供试样品为该6个菌株子实体原基期、幼菇期、采摘期、开伞期4个不同发育阶段的样品,共24个样品。
1.2 试验方法
1.2.1 子实体取样
常规栽培获得的双孢蘑菇子实体按照之前的方法[8]进行不同发育阶段的采样,经液氮速冻后于-80℃保存备用。
1.2.2 总RNA提取
待测子实体样品加液氮研磨成粉末,用真菌总RNA提取试剂盒(Fungal Total RNA Extraction Kit,OMEGA公司产品)按照试剂盒说明书进行总RNA提取。
1.2.3 转录组测序
提取的总RNA送交北京百迈客生物科技有限公司进行转录组测序。包括样品检测、文库构建、上机测序、测序数据处理及质量控制等步骤,均按照之前的方法[10-11]进行。
1.2.4 生物信息学分析
测序数据的生物信息学分析包括与参考基因组的序列比对、可变剪接分析、新基因发掘、差异表达基因识别、差异表达基因功能注释和富集分析等,均按照文献[10]进行。基因表达量计算采用FPKM(reads per kb per million reads)方法[12]。
2 结果与分析
2.1 测序数据产出统计
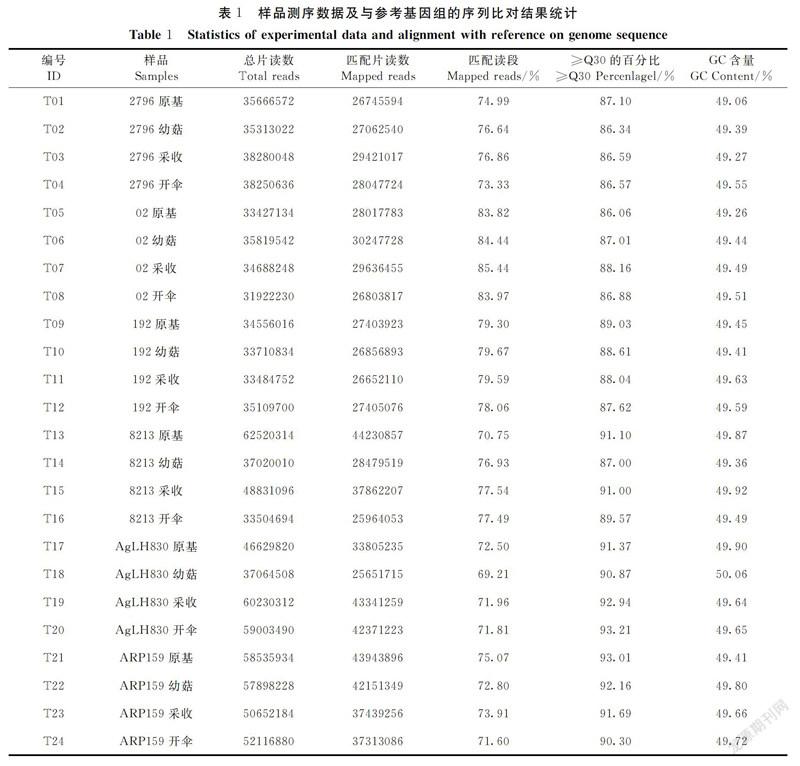
24个样品数据产出统计见表1。经过测序质量控制,共得到128.98 Gb Clean Data,各样品Q30碱基百分比均不小于86.06%。利用TopHat 2[13]软件将Clean Reads与参考基因组进行序列比对,获得测序样品特有的序列特征信息及在参考基因组或基因上的位置信息。根据比对结果,各样品的Reads与双孢蘑菇指定参考基因组的比对效率在69.21%~85.44%。2.2 新基因发掘
选择双孢蘑菇参考基因组序列,使用Cufflinks软件对匹配的Reads进行拼接,并与参考基因组(https://genome.jgi.doe.gov/Agabi_varbisH97_2/Agabi_varbisH97_2.home.html)注释信息进行比较,共发掘677个新基因。使用BLAST[14]软件将发掘的新基因与COG[15],GO[16],KEGG[17],Swiss-Prot[18],NR[19]等数据库进行序列比对,在各数据库获得相应注释信息的新基因数量见表2,总共有237个基因获得注释。
2.3 差異表达分析
根据比对结果计算得到各基因表达量,使用EBSeq[20]进行差异分析,获得测序样品之间包括上调基因和下调基因在内的差异表达基因集,其数目统计见表3。可以看出以原基期为参照,不同菌株在后3个发育阶段中基因上调或下调表达趋势并不一致,以下调表达为主。对差异表达基因进行GO、COG、KEGG、Swiss-Prot、NR等数据库的功能注释,大多数的差异基因获得了相应的注释信息(表4)。
2.4 菌株间共有差异基因分析
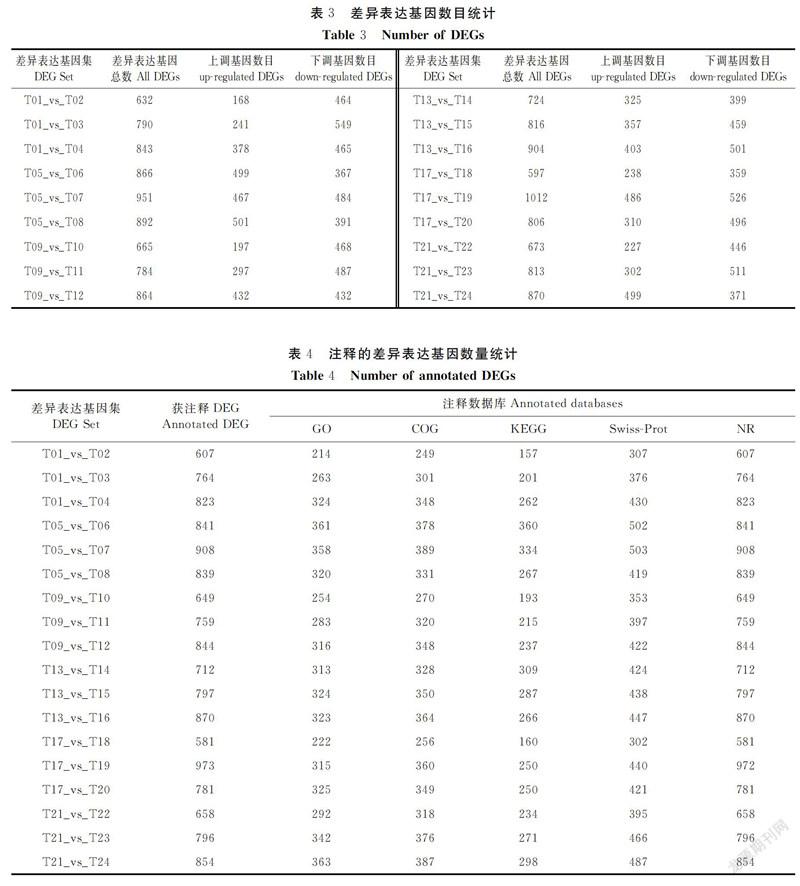
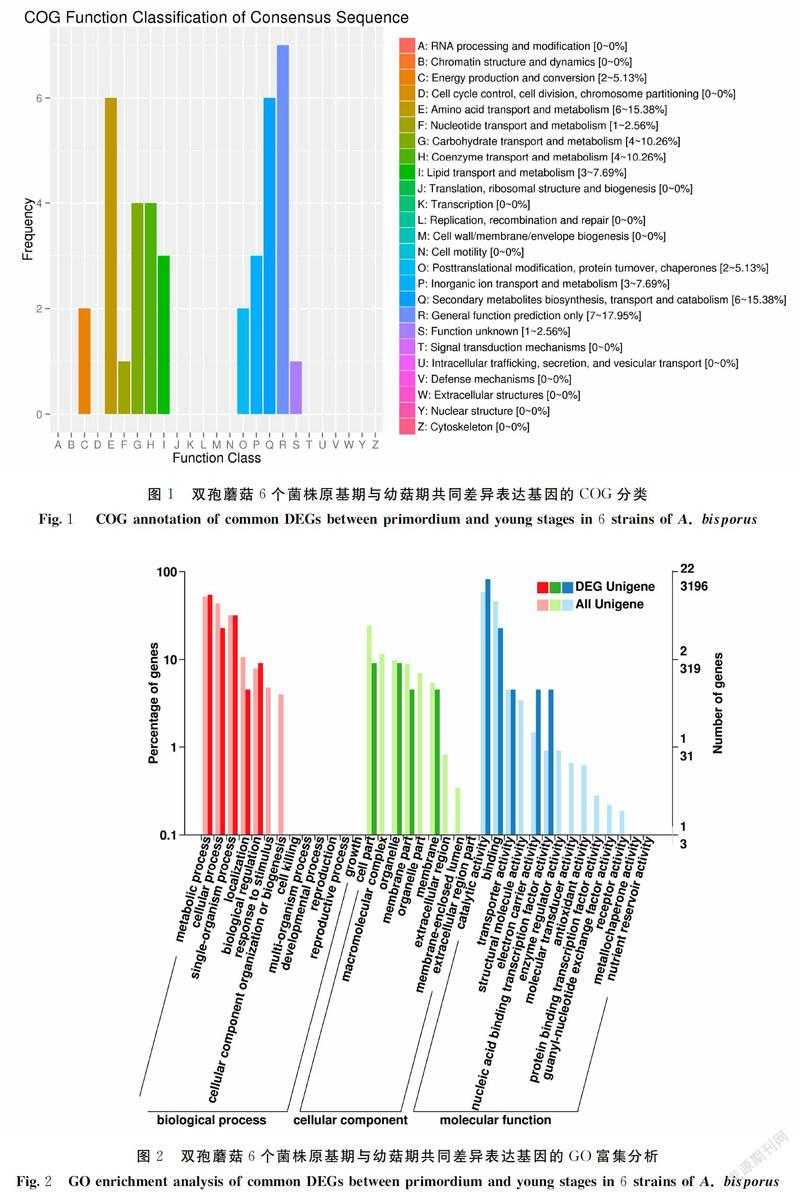
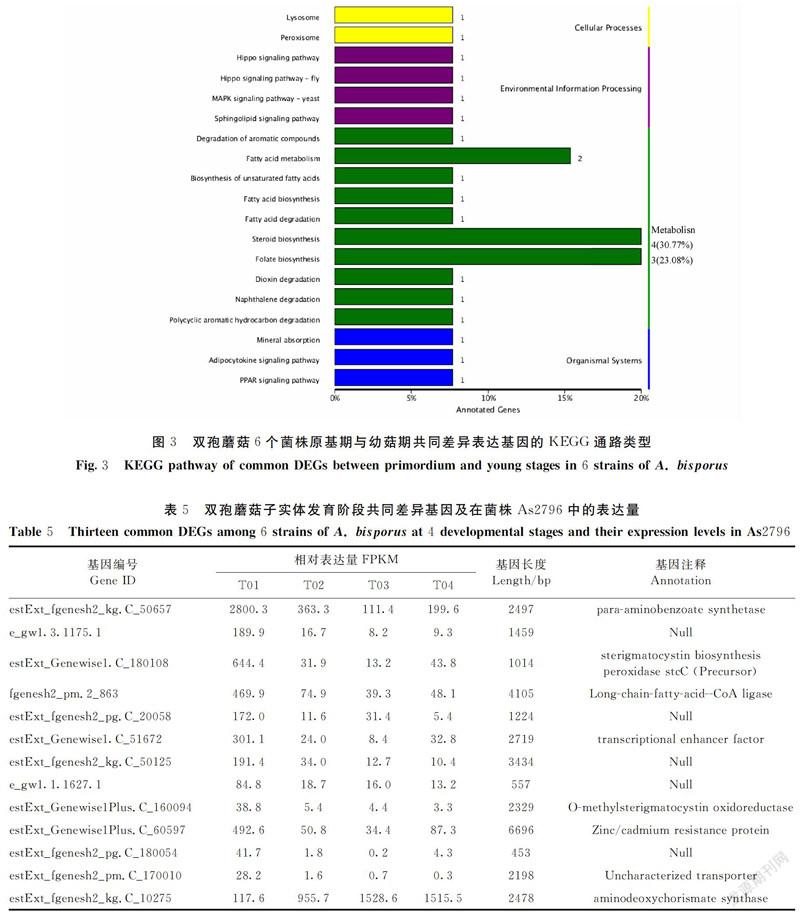
对双孢蘑菇6个菌株子实体4个不同发育阶段的差异表达基因进行分析,发现与原基期相比,6个菌株在幼菇期、采摘期和开伞期分别有49、82、73个共同差异表达基因,并对其进行功能注释和富集分析。图1~3显示了原基期与幼菇期49个共同差异基因的分析结果,其中COG分类注释结果表明差异基因主要集中在氨基酸转运与代谢、次级代谢产物合成、转运与分解等11个功能类别,GO富集结果显示它们主要参与代谢过程、催化活性等14项生物学过程、细胞组分或分子功能,而KEGG富集分析则表明这些差异基因主要参与脂肪酸代谢、类固醇与叶酸生物合成等途径。
进一步统计表明,上述来自6个菌株不同发育阶段的49、82、73个共同差异表达基因中有13个基因是相同的,这些基因与原基期相比的上下调差异表达贯穿于子实体后续阶段的发育过程。表5列出了这些基因的ID、长度及在As2796子实体4个发育阶段中的相对表达量,在其他5个菌株中的表达量趋势也是一致的。与原基期比较,只有1个基因(Gene ID:estExt_fgenesh2_kg.C_10275)是上调表达的,该基因在Swiss-Prot注释为可能是aminodeoxychorismate synthase(氨基脱氧分支酸合成酶)。其余的12个基因在后3个阶段中是下调表达的,在Swiss-Prot数据库中分别注释为transcriptional enhancer factor(转录增强因子) 、Long-chain-fatty-acid-CoA ligase(长链脂肪酸辅酶A连接酶)、O-methylsterigmatocystin oxidoreductase(甲基柄曲霉素氧化还原酶)、Zinc/cadmium resistance protein(锌/钙抗性蛋白)等。
3 讨 论
本次转录组测序共24个样品,即6个代表性双孢蘑菇菌株的子实体原基期、幼菇期、采摘期、开伞期等4个不同发育阶段的样品,通过生物信息学分析及进一步的比较,发现了一批在双孢蘑菇子实体不同发育阶段具有显著差异表达的基因,部分基因的相对表达量差异甚至高达千倍以上。
课题组前期已单独分析了双孢蘑菇As2796子实体发育的转录组,对差异基因进行了注释并筛选出了4个发育阶段中基因相对表达量连续下调至痕量水平以及从痕量水平连续上调表达的差异基因[10]。吴小梅等[21]之前也对双孢蘑菇一个菌株子实体发育的3个时期样品进行了转录组测序分析,列出了3个发育阶段相对表达量大于15倍的18个差异基因。本文使用6个代表性菌株进行子实体发育不同阶段的转录组分析,获得了不同菌株间同个阶段的共同差异基因,以及不同菌株不同阶段的共有差异基因,从上述分析的不同角度对双孢蘑菇子实体发育中重要的差异基因进行了注释与探讨。这些显著的差异表达基因以及在6个菌株不同发育阶段的共同差异表达基因是否与双孢蘑菇生长发育、产量、质量等相关,值得后期进行更深入的筛选与验证分析。
参考文献:
[1]PLAZA D F, LIN C W, van der VELDEN N S J, et al.Comparative transcriptomics of the model mushroom Coprinopsis cinerea reveals tissue-specific armories and a conserved circuitry for sexual development[J]. BMC Genomics, 2014, 15(1):492-509.
[2]TEICHERT I, WOLFF G, KCK U, et al. Combining laser microdissection and RNA-seq to chart the transcriptional landscape of fungal development[J]. BMC Genomics, 2012, 13(1):511-529.
[3]杨芳,许波,李俊俊,等. 鸡枞菌转录组分析揭示其对木质纤维素的降解功能[J]. 微生物学报, 2012, 52(4):466-477.
YANG F, XU B, LI J J, et al. Transcriptome analysis of Termitomyces albuminosus reveals the biodegradation of lignocellulose[J]. Acta Microbiologica Sinica, 2012, 52(4):466-477.(in Chinese)
[4]CHEN L F, GONG Y H, CAI Y L, et al. Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation[J]. PloS One, 2016, 11(8), e0160336.
[5]FU Y P,DAI Y T,YANG C T,et al.Comparative transcriptome analysis identified candidate genes related to Bailinggu mushroom formation and genetic markers for genetic analyses and breeding[J].Scientific Reports, 2017,7(1):9266.
[6]陈美元. 双孢蘑菇子实体原基与菇蕾蛋白质表达变化分析[J],食用菌学报,2012, 19(3):15-20.
CHEN M Y. Differential Expression of Proteins During the Primordium and Button Stages of Agaricus bisporus[J]. Acta Edulis Fungi, 2012, 19(3):15-20.(in Chinese)
[7]陈美元,廖剑华,李洪荣,等. 双孢蘑菇子实体发育后期差异表达蛋白质分析[J],菌物学报,2013, 32(5):855-861.
CHEN M Y, LIAO J H, LI H R, et al. Analysis of differentially expressed proteins in later developing stage fruitbody of Agaricus bisporus[J]. Mycosystema, 2013, 32(5):855-861.(in Chinese)
[8]陈美元,廖剑华,李洪荣,等.双孢蘑菇子实体发育差异蛋白质组分析[J],菌物学报,2015, 34(6):1153-1164.
CHEN M Y, LIAO J H, LI H R, et al. Developmental proteomics analysis of the button mushroom Agaricus bisporus[J]. Mycosystema, 2015, 34(6):1153-1164.(in Chinese)
[9]CHEN M Y, LIAO J H, LI H R, et al. iTRAQ-MS/MS proteomic analysis reveals differentially expressed proteins during post-harvest maturation of the white button mushroom Agaricus bisporus[J]. Current Microbiology, 2017, 74(5):641-649.
[10]施肖堃,蔡志欣,郭仲杰,等. 雙孢蘑菇As2796子实体发育转录组测序分析[J],福建农业学报,2018, 33 (3):282-287.
SHI X K, CAI Z X, GUO Z J, et al. Analysis of Agaricus bisporus Fruitbody Development by Transcriptome Sequencing[J]. Fujian Journal of Agricultural Sciences, 2018, 33 (3):282-287.(in Chinese)
[11]蔡丹凤,蔡志欣,陈美元,等. 茯苓菌落褐变的转录组测序分析[J],广州中医药大学学报,2017, 34(2):245-249.
CAI D F, CAI Z X, CHEN M Y, et al. Analysis of Poria cocos Mycelia Browning by Transcriptome Sequencing[J]. Journal of Guangzhou University of Traditional Chinese Medicine, 2017, 34(2):245-249.(in Chinese)
[12]MORTAZAVI A, WILLIAMS B A, McCUE K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq[J]. Nature Methods, 2008, 5(7):621-628.
[13]KIM D, PERTEA G, TRAPNELL C, et al. TopHat2:accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions[J]. Genome Biology,2013, 14:R36.
[14]ALTSCHUL S F, MADDEN T L, ZHANG J, et al. Gapped BLAST and PSI BLAST:A New Generation of Protein Database Search Programs[J]. Nucleic Acids Research,1997, 25(17):3389-3402.
[15]TATUSOV R L, GALPERIN M Y, NATALE D A. The COG database:a tool for genome scale analysis of protein functions and evolution[J]. Nucleic Acids Research,2000, 28(1):33-36.
[16]ASHBURNER M, BALL C A, BLAKE J A, et al. Gene ontology:tool for the unification of biology[J]. Nature Genetics, 2000, 25(1):25-29.
[17]KOONIN E V, FEDOROVA N D,JACKSON J D,et al.A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes[J].Genome Biology, 2004,5(2):77.
[18]APWEILER R, BAIROCH A, WU C H, et al. UniProt:the universal protein knowledgebase[J]. Nucleic acids research, 2004, 32:115-119.
[19]DENG Y Y, LI J Q, WU S F, et al. Integrated nr Database in Protein Annotation System and Its Localization[J]. Computer Engineering, 2006, 32(5):71-74.
[20]HANSEN K D, WU Z J, IRIZARRY R A, et al. Sequencing technology does not eliminate biological variability[J]. Nature Biotechnology, 2011, 29(7):572-573.
[21]吳小梅,张昕,李南羿. 双孢蘑菇子实体不同发育时期的转录组分析[J]. 菌物学报, 2017, 36(2):193-203.
WU X M, ZHANG X, LI N Y. Transcriptome analysis of Agaricus bisporus fruiting at different stages[J]. Mycosystema, 2017, 36(2):193-203.(in Chinese)
(责任编辑:黄爱萍)
