
不同产地益母草种质HPLC 指纹图谱分析
2019-08-24 06:33邵璐彬徐建中罗页思孙乙铭王志安
浙江中西医结合杂志 2019年8期
邵璐彬 徐建中 罗页思 孙乙铭 王志安
益母草(Leonurus japonicus Houtt)为唇形科一年生或二年生草本植物,具有调经,行血生血、祛瘀、消水、安胎等功效,是中医治疗妇科疾病运用广泛的中药之一,也是妇科要药。益母草药材的主要有效成分是生物碱类,主要为益母草碱(leonurine)、水苏碱(stachydrine)、和益母草啶(leonuridine)、益母草宁(leonurinine)等,在各生物碱中,以水苏碱含量较高,其次是益母草碱[1]。由于益母草传统采收是在花前期,此时益母草碱等生物碱含量非常低,基本上不符合药典规定[2]。而当年播种当年采收的益母草,均不发生抽苔,植株呈“芥菜”型,茎极短,叶片数多,由于益母草生物碱含量主要集中在叶片中,因此植株益母草碱等生物碱含量高,而且不同产地的益母草生物碱成分含量差异较大。本试验主要通过HPLC 色谱指纹图谱技术分析评价不同产地的益母草在生物碱类成分方面的差异性,为益母草优良品种的选育提供科学依据。
1 材料与仪器
Aglient HP1200 高效液相色谱仪(美国安捷伦公司);二极管阵列检测器(DAD)(美国安捷伦公司);AB204-S 型电子天平(Mettler Toledo 公司);C18 柱(250mm×4.6mm,5μm)(北京迪科马科技有限公司);恒温水浴锅(江苏金坛金城国胜实验仪器厂)。盐酸益母草碱对照品111823-201303(中国食品药品检定研究院);无水乙醇(杭州化学试剂有限公司);乙腈(色谱纯)(默克股份有限公司);甲酸(上海麦克林生化科技有限公司)。
供试材料益母草种子来源(见表1),将各益母草种质统一种植在浙江省义乌市和溪村益母草种质资源圃,经浙江省中药研究所徐建中高级工程师鉴定为益母草,取营养生长期未开花鲜益母草,70℃烘干,粉碎(过三号筛)。
2 方法与结果
2.1 色谱条件 色谱柱:C18 柱(250mm×4.6mm,5μm),北京迪科马科技有限公司提供;进样量:20μL;检测波长:277nm;柱温:30℃;流速:1mL/min;流动相:乙腈-0.1%甲酸;流动相梯度洗脱比例见表2。

表2 梯度洗脱流动相比例
2.2 标准品溶液制备 精密称取干燥至恒重的盐酸益母草碱对照品0.0035g,置10mL 容量瓶中,加70%乙醇溶解并稀释至刻度,摇匀,再取1mL 置10mL 容量瓶中,加70%乙醇溶解并稀释至刻度,摇匀制成每1mL 含35μg 的溶液,作为对照品溶液。对照品色谱图见图1。
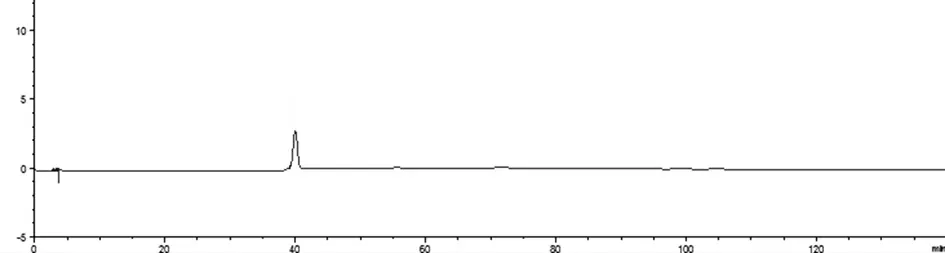
图1 标准品色谱图(盐酸益母草碱)
2.3 供试品溶液制备 精密称取约1g 益母草药材粉末(过三号筛)置具塞平底烧瓶中,精密加入50mL水,称定重量,加热回流3h,补重过滤,精密量取20mL 滤液至分液漏斗中,用乙酸乙酯萃取4 次,收集乙酸乙酯层蒸干,再用70%乙醇定容至10mL,过滤进样。
2.4 方法学考察
2.4.1 色谱指纹峰的标定 以参照物盐酸益母草碱对应的色谱峰的保留时间和峰面积为1,计算其他各色谱指纹峰的相对保留时间及相对峰面积[3]。
2.4.2 精密度试验 精密吸取同一益母草样品溶液,重复进样6 次,按“2.1”项的方法进样,考察各峰相对保留时间、相对峰面积及RSD 值。结果表明共有峰相对保留时间的RSD 在0.31%~1.0%之间,相对峰面积的RSD 在0.78%~2.33%之间,均<5%,表明实验仪器精密度良好。
2.4.3 重现性试验 精密称取同一益母草样品6份,按“2.3”项方法制备供试品,按“2.1”项的方法进样,考察各峰相对保留时间、相对峰面积及RSD 值。结果表明共有峰的相对保留时间的RSD 在0.18%~0.62%之间,相对峰面积的RSD 在1.12%~2.45%之间,均<5%,表明该实验方法重现性良好。
2.4.4 稳定性试验 精密吸取同一益母草样品溶液,室温下放置,在2、4、6、12、24h 按“2.1”项的方法进样测定,考察各峰相对保留时间、相对峰面积及RSD 值。结果表明共有峰相对保留时间的RSD 在0.18%~0.62%之间,相对峰面积的RSD 在1.13%~2.81%之间,均<5%,表明供试品溶液24h 内稳定。
2.4.5 共有指纹峰的标定 采用国家药典委员会“中药色谱指纹图谱相似度评价系统(2004A)”对每个品种的指纹图谱进行分析,得到指纹图谱[4]以及共有模式指纹图谱,见图2、图3。
通过对15 个种质的指纹图谱的分析,发现不同来源的益母草存在差异,如图2 所示,15 个样品一共有24 个共有峰,峰匹配数达10 个以上样品的峰有38 个,其中吉林通化在108min 左右有较强吸收峰,其余种质在该处几乎没有吸收峰,另外,各品种共有峰面积差异也较显著。
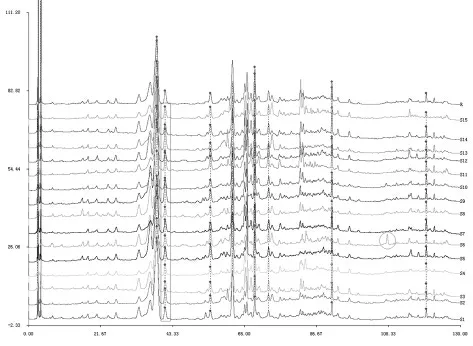
图2 不同品种益母草HPLC 指纹图谱
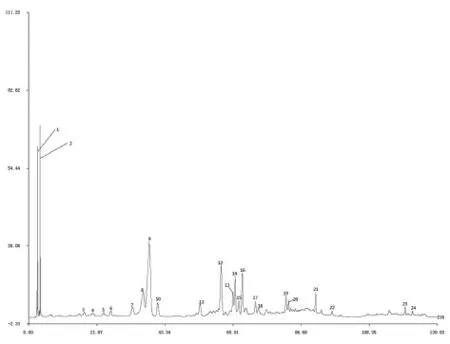
图3 益母草共有模式指纹图谱
2.4.6 相似度分析 采用国家药典委员会“中药色谱指纹图谱相似度评价系统(2004A)”进行数据分析处理,采用多点校正将色谱峰自动匹配,生成对照图谱。将其他样品的色谱峰与对照图谱进行自动匹配,计算S1~S15 样品与对照图谱的相似度,结果表明益母草各种质相似度较高,都在0.92 以上,见表3。
2.4.7 聚类分析 在Aglient 色谱工作站中将指纹图谱的所有峰进行自动积分,以峰面积为聚类依据,用SPSS 统计软件对数据进行聚类统计分析,得到聚类结果,见图4。
如图4 所示,聚类分析结果将15 个种质资源分为四类,第1 类包括9 个种质(湖北孝感、浙江义乌、浙江衢州B、湖南桂东、浙江宁海、吉林通化、江西修水、浙江白龙桥、山东临朐);第2 类包括2 个种质(河南南阳、河南灵宝);第3 类包括3 个种质(浙江衢州A、广西桂林、河南社旗),第4 类为云南陆良种质。
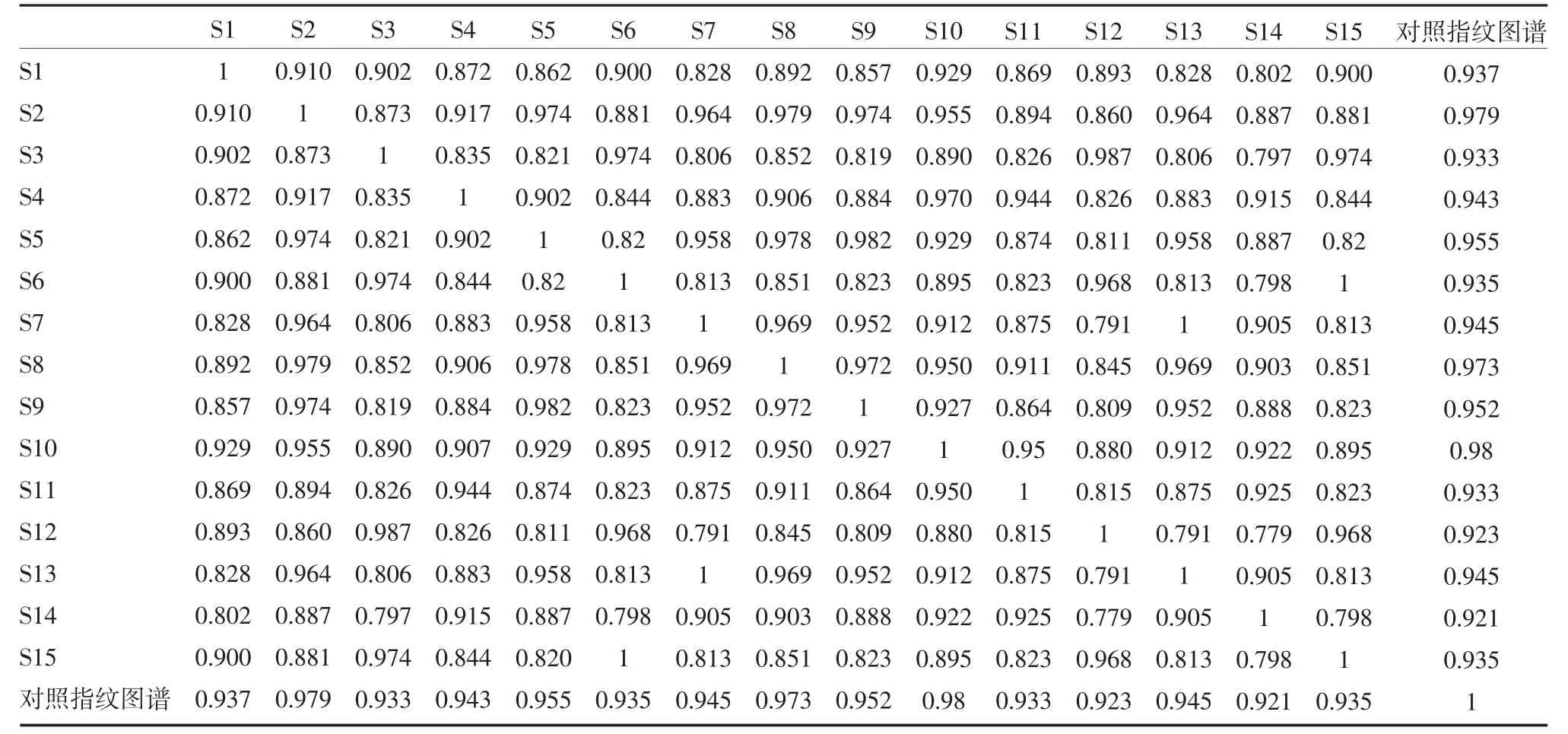
表3 益母草指纹图谱相似度
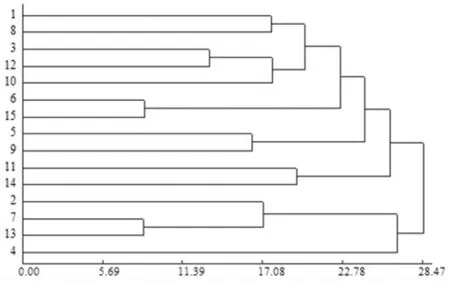
图4 不同益母草品种指纹图谱聚类分析结果
2.4.8 主成分分析 在Aglient 色谱工作站中将指纹图谱的所有峰进行自动积分,以峰面积为聚类依据,用SPSS 统计软件对数据进行PCA 统计分析,得到主成分分析图,见图5。主成分分析的结果基本与聚类分析结果一致,浙江衢州A、广西桂林与河南社旗三个种质可以看作一个区,河南南阳与河南灵宝可以看作一个区,吉林通化与江西修水可以看作一个区,云南陆良与浙江义乌等种质相对其他地区稍远,可以看作单独分类。
3 讨 论
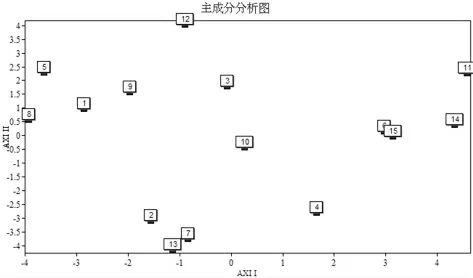
图5 主成分分析图
3.1 提取方法的考察 本实验曾对比研究了水提醇沉法、水提法-过柱纯化法、水提法-有机溶剂萃取法等方法。过柱纯化法选择强酸性阳离子树脂[5]为交换树脂,有机溶剂萃取分别选择氯仿、乙酸乙酯、正己烷三种不同溶剂萃取,将得到的色谱图结果与对照品图谱进行比较,结果显示过柱纯化法与乙酸乙酯萃取法,均能提取出盐酸益母草碱,考虑到实验操作步骤及时间,确定以水提法-乙酸乙酯萃取法为适宜提取方法。
3.2 提取时间和萃取次数的考察 曾分别试验了加热回流时间为1、2、3、4h,并分别用乙酸乙酯萃取5次,收集乙酸乙酯层蒸干,再用70%乙醇定容至10mL,过滤、进样。结果显示:随着提取时间延长,药材中提取的有效成分含量越高,提取3h 与提取4h的含量无较大差异,因此确定回流提取时间为3h。回流后曾试验用乙酸乙酯萃取2、3、4、5 次,收集乙酸乙酯层蒸干,再用70%乙醇定容至10mL,过滤、进样,结果显示:萃取4 次,测得的含量已基本稳定,因此确定萃取次数为4 次。关于萃取:在利用乙酸乙酯萃取时,溶液乳化程度较高,可能是实验环境温度过低,尝试对萃取比例进行调整,发现当乙酸乙酯:水溶液=2:1 时,乳化程度大大减弱。
3.3 色谱条件考察 曾分别试验了237、257、277 和297nm 等四种波长进行测定,结果表明277nm 波长测定时具有较好的紫外吸收和丰富的色谱峰信息,因此确定了以277nm 作为检测波长。还分别考察了20℃、25℃、30℃柱温下的色谱图,发现25℃与30℃色谱峰分离效果较好且无明显差别,考虑柱子因素,确定柱温为30℃。另外,还分别试验了以乙腈-水、乙腈-0.1%甲酸、乙腈-0.1%磷酸等为流动相,结果表明以乙腈-0.1%甲酸梯度系统时各色谱峰分离较好,通过试验确定了适宜的梯度洗脱程序。
3.4 益母草主要有效成分为生物碱类,有文献表明受到物候、光照强度和土壤水分的影响,不同产地益母草药材生物碱类成分含量会有所差异[6-8]。本实验建立了益母草中生物碱类等成分的高效液相色谱指纹图谱,并将15 个不同产地的益母草进行比较,通过实验分析发现,各种质之间化学成分种类相似度较高,说明益母草药材的化学成分种类基本一致;而化学成分含量差异较大,可能是种质之间有差异。
3.5 指纹图谱已成为国际公认的控制中药或天然药物质量的最有效手段[9]。目前对益母草高效液相指纹图谱的研究,多数集中在益母草注射液及益母草胶囊,且多为盐酸水苏碱指纹图谱[10-11]。钟宁远等[11]利用高效液相色谱法建立了益母草流浸膏指纹图谱,得到6 个共有峰,为综合评价和控制产品质量提供依据。本实验通过考察采集于浙江义乌种质资源圃的15 份不同种源益母草药材,构建益母草中盐酸益母草碱高效液相指纹图谱及其共有模式图谱,得到共有峰24 个,可评价益母草药材内在质量及品种的特异性,从而为其资源的现代化开发利用及质量控制提供科学依据。
猜你喜欢
中草药(2022年1期)2022-01-13
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年2期)2021-03-29
婚育与健康(2020年11期)2020-12-23
数码世界(2018年1期)2018-12-23
读与写·下旬刊(2018年6期)2018-07-14
妇女生活(2017年10期)2017-10-10
科学与财富(2017年17期)2017-06-16
化学教学(2015年12期)2015-12-12
中国民族民间医药·下半月(2014年4期)2014-09-26
