
肿瘤坏死因子受体相关周期性发热综合征1例病例报告
2019-08-23 10:58王超颖徐益萍卢美萍
中国循证儿科杂志 2019年3期
王超颖 郭 莉 徐益萍 卢美萍
作者单位 浙江大学医学院附属儿童医院 杭州,310003
1 病例资料
男,8岁4月。因“反复发热、皮疹5年,再发3 d”于2018年8月2日入浙江大学医学院附属儿童医院(我院)。
患儿于3岁左右无明显诱因出现反复发热。每次病程10~20 d,热峰39~40℃,每天出现2次热峰,服用退热药后1~2 h体温可降至正常。发热前常伴寒战。发热时偶有皮疹,多于发热1周时出现,为分布于胸、腹部的淡红色斑丘疹,压之褪色,不伴瘙痒,无痛感,热退后皮疹好转。发热时偶有乏力,热退后好转。偶伴口腔溃疡、咳嗽、流涕。无症状间歇期时间不固定,每年发热1~4次,季节交替时多见。4岁时高热惊厥1次。曾于2018年3月住院期间镜下血尿(RBC10~31个/HP)平素无腹痛、腹泻,无尿频、尿急和尿痛,无泡沫尿,无头痛、胸痛、关节肿痛、肌痛,无眶周水肿、双眼充血。3年来先后5次因“发热待查”入住我院,曾诊断“急性支气管肺炎、脓毒症、原发性免疫缺陷病?中度贫血、血尿、幼年特发性关节炎(全身型)?”等,予哌拉西林他唑巴坦、阿奇霉素、万古霉素等抗感染治疗,好转后出院。3 d前再次出现发热,热型同上,无皮疹。
既往史、出生史、喂养史和家族史均无异常描述。
生长发育史:17个月会走路,24个月会叫爸爸、妈妈。
预防接种史:接种卡介苗后出现“左锁骨下淋巴结炎”,淋巴结渐增大,6月龄时行“引流术”后好转;偶有延迟接种疫苗,但无遗漏。
体格检查:T 37.0℃,P 120·min-1,R 24·min-1,BP 96/65 mmHg,体重20.5 kg(年龄别体重Z评分-1.83),身高121 cm(年龄别身高Z评分-1.39)。神志清楚,反应可,全身未见皮疹,浅表淋巴结未及肿大。双侧扁桃体Ⅰ°肿大,未见渗出。心、肺、腹、四肢关节、神经系统查体未见异常。
实验室检查:血WBC 14.1×109·L-1,N 0.71,Hb 102 g·L-1,PLT 400×109·L-1,CRP 82.7 mg·L-1,ESR 73 mm·h-1;尿常规、肝肾功能、心肌酶谱、铁蛋白和降钙素原均未见异常;免疫球蛋白、淋巴细胞亚群、甲状腺功能、类风湿因子、抗核抗体和抗中性粒细胞胞质抗体检测均未见异常。
采用流式细胞微球芯片技术捕获人T辅助细胞1/2细胞因子试剂盒Ⅱ(美国BD公司)和FACSCalibur流式细胞仪(美国BD公司)检测细胞因子。本次住院IL-2、IL-4、IL-6、IL-10、TNF-α和IFN-γ分别为1、1、19.8、1、1和1 pg·mL-1。既往多次住院细胞因子检测情况:IL-6明显升高,IL-10正常或轻度升高,TNF-α正常,IL-2、IL-4、INF-γ正常或降低。
病原学检查:EBV-DNA、EBV抗体、CMV-DNA、CMV-抗体、TORCH、乙肝、HIV、梅毒、丙肝-抗体、PPD试验、T-SPOT、肥达反应、血培养、骨髓培养、抗链球菌溶血素“O”检测均未见异常。脑脊液常规、生化、培养未见异常。
遗传代谢和血液肿瘤检测:血串联质谱和肿瘤标志物未见异常,骨髓细胞学检查显示,粒系增生显著活跃。
影像学检查:X线胸片、心电图、腹部B超、心脏彩超、头颅MRI平扫均未见异常。
取得患儿家长知情同意后,分别取患儿及其父母外周血2 mL,提取基因组DNA。针对患儿基因组DNA,使用标准文库构建试剂盒(北京迈基诺基因科技股份有限公司)进行基因组文库构建。采用目标序列捕获探针(北京迈基诺基因科技股份有限公司)对免疫系统相关的269个候选基因外显子区域进行捕获,获得目标基因富集文库。借助Illumina HiSeq X10测序平台对富集文库进行高通量测序,获得原始测序数据。将原始测序数据去除污染和接头序列,利用BWA软件将过滤后序列与NCBI数据库人类基因组参考序列(hg19)比对,用GATK软件分析得出单核苷酸变异(SNV)和插入缺失突变(INDEL)的相关信息,ANNOVAR软件注释,筛选分析基因突变信息。用PCR和Sanger测序验证经分析筛选后得到的变异位点,在患儿、父亲和母亲中进行共分离验证。图1显示,患儿TNFRSF1A基因有1个杂合突变c295T>C(p.C99R),患儿父母该位点无变异,该突变为新发突变。
诊疗经过:结合患儿病史,肿瘤坏死因子受体相关周期性发热综合征 (TRAPS)诊断明确。入院后予依那西普25mg(1.2 mg·kg-1)皮下注射后当天体温降至正常;出院后予依那西普25 mg/12.5 mg,每周1次(25 mg和12.5 mg交替使用,剂量相当于每周0.9 mg·kg-1)皮下注射,随访6个月,未再出现发热等症状,随访血常规、肝肾功能和ESR未见异常,细胞因子IL-2、IL-4、IL-6、IL-10、TNF-α和IFN-γ分别为25、1、4.8、1.6、35.3和1 pg·mL-1。
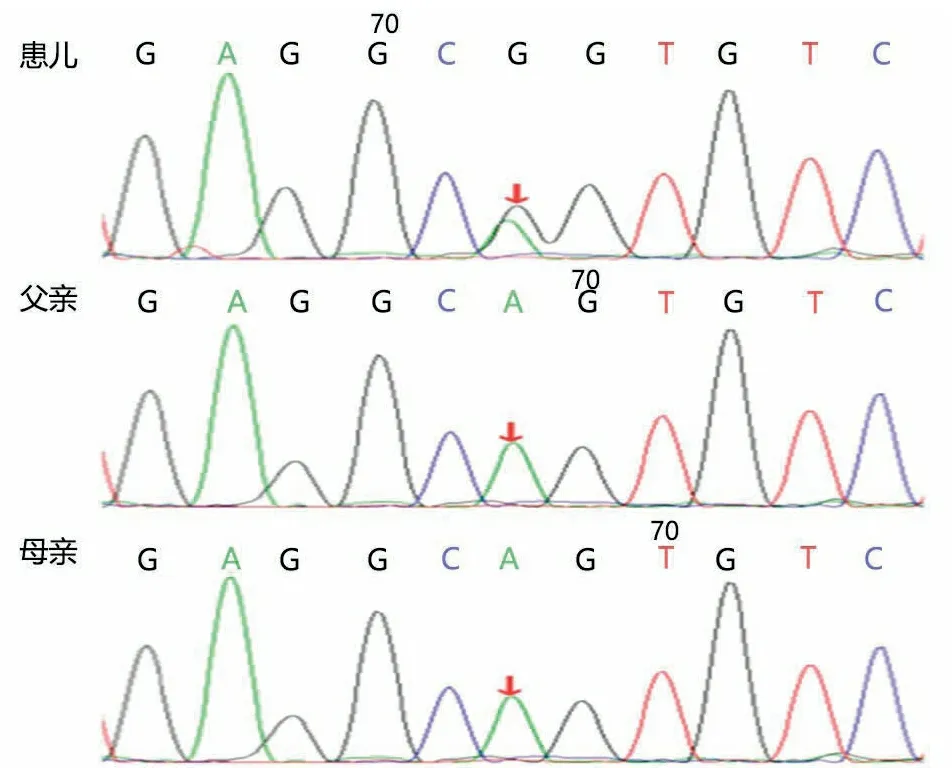
图1 家系Sanger测序结果
注 患儿TNFRSF1A基因存在杂合突变c295T>C
2 讨论
TRAPS为自身炎症性疾病,发病率约1/1 000 000[1],亚洲人发病少见,国内鲜有报道。1982年Williamson等[2]在1个爱尔兰家族中首次发现TRAPS,故曾称其为家族性爱尔兰热。TRAPS为常染色体显性遗传,突变基因定位于12p13,编码肿瘤坏死因子受体1A(TNFRSF1A或TNFR1)[3]。发病机制尚不明确。TNFR1由金属蛋白酶介导,从细胞膜上脱落,游离的受体可竞争性结合TNF,有研究认为,TNFR1基因突变导致TNFR1脱落减少,游离的TNFR1结合TNF减少,更多的TNF与细胞表面受体结合,进而导致下游NF-κb等信号通路长期活化[4]。另外,错误折叠的TNFR1滞留在内质网中,导致TNF诱导的细胞凋亡减少,对固有免疫刺激的敏感性增加,分泌细胞因子IL-1β、TNF-α、IL-6等增多[5]。
TRAPS多于10岁前发病(中位年龄4.3岁),约10%患者于30岁后发病,男女比例约为1∶1。临床表现主要有发热、皮疹、肌痛、结膜炎、眶周水肿、腹痛、胸痛、关节痛或关节炎等,可由应激或外伤等诱发。发热及伴随症状一般持续至少5 d,常持续超过2周[6]。10%~15%患者可继发淀粉样变性,主要发生于肾脏,也可发生于肝脏、甲状腺及其他器官[7]。其他少见的临床表现包括心包炎、胸膜炎、睾丸炎等[8]。实验室检查可有ESR增快,CRP和血清淀粉样蛋白(SAA)增高,其中SAA是与肾脏淀粉样变性相关的早期指标。在疾病缓解期,以上指标可正常或轻度升高。患者自身抗体通常阴性或低滴度阳性[9]。确诊依靠基因诊断,但一些散发病例可无明确的致病基因突变。
目前关于TRAPS有两项大规模的临床研究。2013年儿童风湿病国际试验组织[1]分析了158例TRAPS患者的临床资料,最常见的TNFR1突变为R92Q(34%)和T50M(10%);R92Q突变患儿起病年龄稍迟(中位年龄5.7岁),皮疹和眼部症状轻微,但头痛更常见。2016年日本一项研究[6]统计了169例TRAPS病例,最常见突变为T61I(49%),该突变也可出现在正常人群中,未检测到R96Q和P46L突变。与欧洲人群相比较,日本患者发热的发生率更高,腹痛和淀粉样变性的发生率更低。目前国内仅有1例TRAPS报告[10],为14岁男孩,起病10余年,以发热、皮疹、关节痛和肌肉痛为主要临床表现,检测到TNFR1基因C58C/R突变,其祖父和父亲也检测到同一突变,加用塞来昔布口服后好转。
本文患儿3岁起病,主要表现为发热、皮疹,热程10~20 d,无其他伴随症状,可能与季节交替导致的气温变化诱导发病。急性期WBC、CRP升高,ESR增快。本次住院细胞因子变化主要为IL-6升高,既往多次住院也以IL-6升高明显、IL-10正常或轻度升高。本次住院使用依那西普治疗后,体温正常时复查细胞因子,IL-6降至正常,提示IL-6在该病发病机制中可能起一定作用。患儿TNFR1基因检测到1个新发杂合突变C99R,既往已有报道该突变与TRAPS有关[11]。结合病史和基因检测,TRAPS诊断明确。本例TRAPS临床诊断评分[12]66分(热程>6 d,19分;迁移性皮疹,18分;无呕吐,14分;无阿弗他溃疡,15分),大于43分界值。使用依那西普有效,随访半年未再出现发热症状,炎症指标正常,贫血改善。
TRAPS的治疗目标主要为控制症状、减少发作及淀粉样变性风险。NSAIDs对于缓解发热等症状可能有效。短期应用糖皮质激素可有效终止发作,但随使用时间延长,可能出现激素耐药。对于频繁发作和(或)在发作间期存在亚临床炎症的患者,推荐使用IL-1受体拮抗剂或依那西普维持治疗,以减少激素的使用[13,14]。依那西普对部分患者有效,但作用可随时间衰减。不建议使用抗TNF单克隆抗体(如英夫利昔单抗、阿达木单抗)治疗[13],因其可能导致症状加重。IL-1受体拮抗剂如阿那白滞素和卡那单抗等对大多数患者有效[15,16]。此外,也有IL-6受体拮抗剂(托珠单抗)成功应用的报道[17]。
由于本病少见,发热、皮疹症状及炎症指标增高均为非特异性,因此易被误诊为脓毒血症、全身型幼年特发性关节炎等,诊断较为困难,导致大量抗生素应用,长期慢性炎症导致贫血、生长发育受限等,临床上应引起重视。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
昆明医科大学学报(2021年12期)2021-12-30
现代临床医学(2021年4期)2021-07-31
科学导报(2021年29期)2021-06-03
中国生殖健康(2020年4期)2021-01-18
科海故事博览·下旬刊(2019年6期)2019-04-16
医学研究杂志(2015年12期)2015-06-10
右江医学(2014年1期)2014-03-22
疯狂英语·口语版(2013年8期)2013-08-19
为了孩子(孕0~3岁)(2001年24期)2001-01-23
