
保健品中原花青素含量检测方法的比较研究
2019-07-31 06:36汪玉玲陈星蓉陆婷婷林腾奕梁培荣
质量安全与检验检测 2019年3期
汪玉玲 陈星蓉 陆婷婷 李 桑 林腾奕 梁培荣
(广东省食品检验所 广东广州 510435)
1 前言
原花青素(Procyanidins)是植物中一大类由儿茶素、表儿茶素、没食子酸和表儿茶素没食子酸酯等单体键合而成的聚多酚化合物的总称[1],是一种应用前景非常广阔的新型植物性抗氧化剂,能强效地清除人体自由基。研究表明[2],原花青素在体内抗氧化、清除自由基能力是维生素C 的20 倍、维生素E的50 倍,能防治由自由基引发的80 多种疾病,有预防心脏病、保护大脑神经系统[3]、降低血脂、抗肿瘤[4]、抗氧化(延缓衰老)[5]、调节免疫力[6]及保护视力等作用。近年来,以葡萄籽为提取原料的原花青素已广泛用于食品、药品、保健品及化妆品领域中,我国对含原花青素的保健品也进行了大量的研制开发和应用,市场发展迅猛,但产品质量参差不齐。因此,建立一种准确高效的测定保健品中原花青素含量的方法具有十分重要的意义。
目前,我国对保健品中原花青素的检验是用铁盐催化比色法。其测定原理是原花青素在加热的酸性条件且和铁盐催化作用下,C-C 键断裂而生成深红色花青素离子(即氰定),测定在特征波长525 nm处的吸光度。但在实际操作中发现,样品采用沸水浴回流加热,操作烦琐,易受氧气[7]、日照、温度等因素的影响,平行性较差,不适于日常大批次的保健品中原花青素的检测;且比色法分离能力差,不能较好地排除其他有色杂质对待测物的影响,测定结果的准确度较低。因此,对于保健品中原花青素含量的测定是目前面临的一个难题。
在已有的相关文献中,对原花青素的检测方法主要有铁盐催化比色法[8]、香草醛-盐酸法[9]、正丁醇-盐酸法[10]、高铁盐-铁氰化钾比色法[11]、紫外分光光度法[12]等,这些方法都要求反应中的特征性显色明显,且显色稳定时间短,不能对相似的杂质成分进行分离测定,较适于测定原花青素纯度高的产品,对一般原料中原花青素的测定分离能力还不够理想,准确度和灵敏度较低。为此,本文在铁盐催化比色法的基础上,优化了前处理过程,结合高效液相色谱法的高选择性和高准确度,建立了一种适用于分析保健食品中原花青素含量的铁盐催化-高效液相色谱法[13,14],该方法前处理较简便、重现性好,可以有效地分离待测样中共存的其他有色杂质,避免干扰,有较高的准确度和灵敏度,适于实验室大批次的检验工作,可作为快速测定保健食品中原花青素含量的一种参考方法,为保健品的质量监管提供技术支撑。
2 材料与方法
2.1 材料与试剂
原花青素标准品(纯度≥95.2%,上海源叶生物科技有限公司);甲醇(色谱纯,美国默克公司);甲酸(色谱级,上海阿拉丁生化科技有限公司);异丙醇(色谱纯,美国BCR 公司);二氯甲烷(色谱纯,上海麦克林生化科技有限公司);正丁醇(分析纯,广州化学试剂厂);盐酸(分析纯,广州化学试剂厂);硫酸铁铵(分析纯,广州化学试剂厂);超纯水由实验室制备。
实验样品:原花青素葡萄籽软胶囊、原花青素葡萄籽精华片、原花青素桑葚黑枸杞果汁口服液(均为市售常见商品)。
2.2 仪器与设备
Prominence UFLCXR 高效液相色谱仪、SPD-M20A检测器(日本岛津公司);UV-2700 紫外/可见分光光度计(日本岛津公司);Milli-Q Reference 超纯水仪(美国密理博公司);HWS28 型电热恒温水浴锅(上海一恒科学仪器有限公司);电热鼓风干燥箱(上海一恒科学仪器有限公司);KQ-500DE 型台式数控超声波清洗器(昆山市超声仪器有限公司);MS105DU万分之一天平、PL602E 电子天平(瑞士梅特勒公司);20 mL 棕色顶空瓶(浙江欧尔赛斯科技有限公司);0.22 μm 针孔滤膜(德国波尔公司)。
2.3 实验方法
2.3.1 样品制备及提取
(1)片剂:将试样研磨成粉状后,称取1 g(精确至 0.001 g)于 100 mL 容量瓶中,加入 50 mL 甲醇,超声处理20 min,放冷至室温后,加甲醇至刻度,摇匀,离心至澄清后取上清液备用。
(2)胶囊:挤出胶囊内容物,搅拌均匀,称取50 mg(精确至0.001 g)试样置于小烧杯中,用少量二氯甲烷使试样溶解,并洗入200 mL 棕色容量瓶中,加甲醇至刻度,摇匀。
(3)口服液:摇匀后取 1 g(精确至 0.001 g)试样置于25 mL 棕色容量瓶中,加甲醇至刻度,摇匀。
2.3.2 水解反应
取正丁醇:盐酸(95:5,V/V)混合溶液 6 mL 置于20 mL 空瓶中,再加入0.2 mL 硫酸铁铵溶液(称取2 g硫酸铁铵,用浓度为2 mol/L 盐酸溶解,定容至100 mL)和样品溶液1 mL,加盖密封,混匀,置100℃烘箱中加热40 min,然后立即置冰水中冷却,过0.22 μm 滤膜,备用。
2.3.3 标准曲线制备
原花青素标准品经纯度换算后精确称取0.052 9 g,用甲醇溶解并转移到50 mL 棕色容量瓶中,定容到刻度,配制成质量浓度为1.00 mg/mL 的标准溶液,现配现用。用移液枪分别准确移取 50 μL、125 μL,250 μL,500 μL,750 μL,1 500 μL 标准溶液于 5 mL棕色容量瓶中,用甲醇至刻度,摇匀。各取1 mL 用以上步骤进行水解,并制备工作曲线。
2.3.4 色谱条件
色谱柱:Agllent ZORBAX SB-C18 StableBond Analytical (4.6 mm×250 mm,5 μm);流动相:水+甲醇+异丙醇+甲酸=65+22+5+8(V/V);流速:1 mL/min;柱温:30℃;进样体积:10 μL;检测波长:525 nm。
3 结果与分析
3.1 加热方法的优化
利用沸水浴回流加热40 min 后测定, 但实际操作中发现,沸水浴回流操作烦琐,较难适用于实验室大批量的样品检测。针对此问题,本实验将沸水浴回流改进成如下2 种方法:(1)改用25 mL 具塞比色管,于 100°C 沸水浴加热 40 min 反应;(2)改用 20 mL 的棕色顶空瓶,放入 100°C 烘箱加热 40 min。具体操作:选用原花青素葡萄籽软胶囊作为试样,分为低、中、高3 组,分别加入约50%、100%、200%试样含量的标准溶液,同时考察沸水浴回流、直接沸水浴加热及烘箱加热3 种反应方式下的原花青素含量,重复实验3 次(n=3)。不同加热方法的比较结果详见表1。

表1 不同加热方法的比较
由表1可看出,沸水浴回流和直接沸水浴加热这2 种方式测得的样品含量相差不大,但中、高浓度组的加标回收率较低,与烘箱加热相比,后者的试样测定值明显高于前两者,且3 个水平的加标回收率都优于前两者(97.7%~102.6%),相对标准偏差(0.5%~2.3%)也满足理化检验要求,故采用烘箱100°C 加热的方式较优。其原因可能与采用密封的棕色顶空瓶和相对密闭的烘箱环境有关,此操作可有效地隔绝反应过程中氧气、光照对原花青素的氧化,减少实验过程的损失,提高加标回收率,故将样品前处理的加热方法优化为用顶空瓶于烘箱加热。
3.2 水解反应的温度选择
据相关文献表明[15],原花青素的水解反应需要较高的温度,室温下反应速度极慢,在不同水解温度下,花青素离子的生成量差异很大,且该差异不能通过延长加热时间来弥补。为探索原花青素最佳的水解温度,本文将含量为100 mg/L 的原花青素标准溶液置于棕色顶空瓶中密封后在在烘箱中加热,水解反应 40 min,分别对比在 80℃、90℃、100℃、110℃、120℃下5 种烘箱温度下原花青素的测定值。详见图1。
由图1可知,当温度低于100℃时,原花青素的水解反应不完全,随着温度的升高,水解反应速度加快,花青素离子的生成量逐渐增加;当温度在100℃~110℃时,花青素离子的生成量达到最大值;当温度高于110℃,花青素离子的生成量开始有所下降。
其可能的原因是:不同温度会影响不同聚合度的原花青素的C-C 键的断裂程度。随着温度的升高,原花青素单元间的连接键更易断裂,使得花青素离子的生成量增加;但原花青素极不稳定,在高温下又极易失去活性,发生自氧化的现象[16],从而使花青素离子的生成量降低。故综合考虑,本试验采用100℃作为原花青素的最佳水解温度。

图1 不同温度下原花青素含量的变化图
3.3 水解反应时间的选择
为了探索最佳的水解反应时间,本试验将已知含量的原花青素试样置于棕色顶空瓶中密封后于100℃烘箱中加热,分别比较加热反应20 min、40 min、60 min和80 min 时原花青素的测定值。详见图2。
由图2可知,加热约40 min,测得的试样中原花青素含量达到最大值;40 min 后,随着加热时间的延长,结果测定值稍有下降。可能的原因是:高温条件下,原花青素发生自氧化现象,随时间的延长原花青素不断降解,导致含量不断减少,故本试验采取水解反应40 min。

图2 不同水解反应时间下原花青素含量的变化图
3.4 硫酸铁铵催化剂用量的选择
原花青素在加热的酸性条件下氧化为花青素离子,是一种氧化还原反应。早期有人提出,Fe3+可能通过与花青素离子络合作用, 增大吸光系统。后来Lawrence 等[17]证实,在原花青素生成花青素的过程中, Fe3+起到催化剂的作用,可加快反应速率。本文为了考察硫酸铁铵催化剂的最佳用量,取已知含量的原花青素软胶囊为试样,分别对比了在不加、加1%、加2%及加3%硫酸铁铵溶液4 种情况下测得的试样中原花青素的含量。结果详见图3。

图3 催化剂用量对原花青素测定的影响
由图3可知,不加硫酸铁铵溶液的试样水解后颜色明显比加硫酸铁铵溶液的颜色浅,同一样品不加硫酸铁铵溶液时测定值最低,加2%硫酸铁铵溶液时,测定值最高,之后增加用量,测定值都趋于稳定。故采用2%硫酸铁铵溶液做催化剂,既节省试剂的用量又可使水解反应更完全。
3.5 检测方法的比较
3.5.1 色谱法与比色法准确度的比较
目前对原花青素的检测采用铁盐催化比色法。鉴于高效液相色谱法具有较强的分离能力、较好的定性和定量效果,故本试验尝试使用高效液相色谱法进行分析(图4)。比对时采用相同的样品、试剂和优化后完全一致的前处理方法,对处理好的样品分别使用铁盐催化比色法和色谱法进行分析测定。
比较发现:2 种方法的平行性和加标回收率都满足要求,但当试样为含油软胶囊时,铁盐催化比色法测定结果高于色谱法,结果详见表2。且由图4可看到,目标峰原花青素的右侧有一较小的杂质峰,且该杂质峰在525 nm 波长附近同样有较大吸收(图5)。故可能是由于比色法没有分离能力,当原花青素试样成分复杂时,部分有色杂质与待测物混在一起干扰测定,从而使比色法的测定结果偏高。故采用高效液相色谱法测定保健品中原花青素的含量结果更为准确。

表2 2 种方法测定结果的比较

图4 试样在色谱法中分离的色谱图
3.5.2 稳定性试验的比较
取已知含量的原花青素试样加入相同的试剂,在相同条件下进行水解反应,对处理好的试样每隔1 h 分别采用铁盐催化比色法和色谱法进行分析测定,观察样品测定值随时间变化的规律,结果详见图6。
由图6(a)、(b)可知,铁盐催化比色法在试样溶液显色2 h 内测定吸光度值较稳定;而高效液相色谱法在上机的5 h 内,测定结果都基本稳定,含量无显著变化,(相对标准偏差)RSD 为0.5%。
因此,综合考虑,本试验采用高效液相色谱法进行分析测定,该方法相比于铁盐催化比色法,检测结果准确度更高,稳定性更好。

图5 目标峰和杂质峰的最大吸收波长光谱图

图6 测定值随时间的变化趋势
3.6 方法线性范围及检出限、定量限
将原花青素系列工作溶液(10 mg/L、25 mg/L、50 mg/L、100 mg/L、150 mg/L、300 mg/L)按优化后的实验条件进行水解反应,按上述色谱条件进样分析,得到原花青素的色谱图,原花青素标准品的浓度和对应的峰面积详见表3,两者的线性关系详见图7。

表3 原花青素标准品的浓度及峰面积
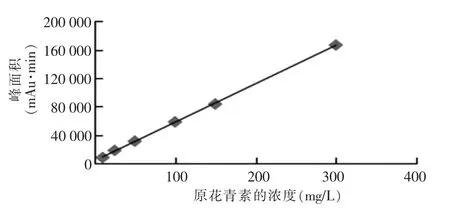
图7 原花青素的标准曲线图
由图7可知,原花青素在浓度为10~300 mg/L时线性良好,线性回归方程为Y=540.7X+3 694.23,相关系数平方为r2=0.999 7。原花青素峰型良好,保留时间为4.147 min,理论塔板数为9 613。
以已知原花青素含量的试样为实验对象,按优化后的方法进行前处理,考察其检出限,以目标化合物色谱峰3 倍信噪比(S/N)计算方法检出限(Limit of Detection, LOD),10 倍信噪比计算方法定量限(Limit of Quantization, LOQ),得到原花青素的方法检出限为1.5 mg/kg,方法定量限为5.0 mg/kg。
3.7 方法的精密度和准确度
分别以原花青素葡萄籽软胶囊、原花青素葡萄籽精华片、原花青素桑葚黑枸杞果汁口服液为试验对象,各分为低、中、高3 组,每组分别加入约50%、100%、200%试样含量的标准溶液,按照本方法优化好的条件进行测定,每组重复试验6 次(n=6),以加标回收率考察本方法的准确度,以相对标准偏差考察方法的精密度,结果详见表4。由表4可知,3 种类型保健品中原花青素的加标回收率为99.0%~103.6%,相对标准偏差(RSD)均小于2.2%,符合理化检验分析方法要求,故本试验所建立的分析保健品中原花青素含量的方法具有可靠的准确度和精密度。

表4 回收率和精密度实验结果
4 讨论
与以前的保健食品中原花青素的测定方法比较,本试验具有以下优势:(1)在加热方式的选择上,采用顶空瓶于烘箱加热代替沸水浴回流加热简化了烦琐的样品前处理过程,适于实验室大批量样品的快速检验,节约时间,可提高检验效率;同时,密闭的烘箱能有效地隔绝氧气和光照,减少氧化,提高测定的准确性、平行性;(2)样品前处理上,在铁盐催化剂的用量、加热温度和水解时间3 方面做了对比研究,结合原花青素的性质,确定了最适原花青素反应的条件,有助于提高测定的准确度,且为今后探索不同资源的原花青素的组分、结构、性质提供了参考价值;(3)在分析方法的选择上,采用高效液相色谱法代替铁盐催化比色法,提高了分离能力,可排除部分有色杂质对待测物的干扰,且灵敏度、精密度、准确度和稳定性都较好,满足检验需求,可作为快速检测保健食品中原花青素含量的参考方法。
近年来,由于原花青素对人体有多种特殊功效,含原花青素的保健品已不断被研究开发利用,市场需求旺盛,与此同时也出现相关质量问题,目前我国关于原花青素的测定方法也尚无统一的标准,故本文所优化的快速准确测定保健品中原花青素含量的方法对日常保健品的质量监管有一定的指导意义和应用推广价值。
猜你喜欢
当代水产(2022年4期)2022-06-05
口腔护理用品工业(2021年4期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
纺织机械(2020年5期)2020-12-14
分析化学(2018年12期)2018-01-22
湖北农业科学(2016年21期)2017-03-18
分析化学(2014年9期)2014-09-26
印刷技术·数字印艺(2014年3期)2014-06-10
筑路机械与施工机械化(2014年4期)2014-03-01
