
Chemotherapy-induced immunological breast cancer dormancy: a new function for old drugs?
2019-07-29 08:43:50SanamPeyvandiQiangLanGiriecaLorussoCurzioegg
Sanam Peyvandi, Qiang Lan, Girieca Lorusso, Curzio Rüegg
1Department of Oncology, Microbiology and Immunology, Faculty of Science and Medicine, University of Fribourg, Fribourg 1700, Switzerland.
2Developmental Biology Program, Institute of Biotechnology, University of Helsinki, Helsinki 00014, Finland.
#These authors contributed equally to the manuscript.
Abstract Breast cancer remains the main cause of cancer-related mortality for women world-wide. Main cause of death is the development of therapy-resistant metastases. Relapses occur with a bimodal temporal distribution, with a first peak at 1-2 years after initial therapy and a second peak 2-3 years later. This discontinuous growth kinetics is consistent with the notion that disseminated cancer cells can remain dormant over a prolonged period of time before resuming growth. How cancer cells enter, sustain and exit dormancy, are unanswered questions with relevance to cancer biology, monitoring and therapy. Investigating mechanisms of breast cancer dormancy remains challenging, as in patients the condition is elusive and experimentally there are only a few models that recapitulate the clinical condition. Thus, developing new models to identify clinically relevant mechanisms and candidate therapeutic targets may open new avenues for novel therapies to induce and prolong dormancy. We have observed that cells surviving chemotherapy can enter a state of immunological dormancy. Using this model, we identified IRF-7/Interferon type I/IFNRA as signaling axis essential for this effect. Here we will review concepts and recent developments in cancer metastasis and dormancy with emphasis on breast cancer, and elaborate strategies to exploit them therapeutically.
Keywords: Breast cancer, chemotherapy, resistance, dormancy, T lymphocytes, interferon
INTRODUCTION
With a few exceptions, as in the case of brain or liver cancer, the main cause of cancer-related death is not the primary tumor itself but rather the formation of secondary tumors, so called metastases, in vital organs, in particular lung, liver, bone or brain, leading to organ destruction and failure, resistance to therapy and cachexia[1,2]. Metastasis formation is believed to be a highly inefficient process[3]. The main rate limiting step in the metastatic process appears to be the ability of tumor cells to adapt to the new environment[4,5]. During this adaptation step at the metastatic site, cells have to establish bidirectional paracrine communication with a new tissue different from the primary site, acquire novel survival capacities and escape immune destruction. While cell autonomous processes, such as genetic evolution and epigenetic modifications, altered gene expression and metabolic adaptation, are essential to the metastatic process, the microenvironments of the primary tumor and of the metastatic site are equally critical determinants of metastasis formation[5-8]. Tumor angiogenesis, the remodeling of the extracellular matrix, the activation of local resident cells and the recruitment of inflammatory cells provide essential contributions to the metastatic process, including in breast cancer[9-11]. At diagnosis, only a minority of cancers have already formed clinically overt metastases (i.e., stage IV)[12]. Those that progress to metastatic disease can do with strikingly different kinetic. For example, lung and colorectal cancers mostly relapse within 1-3 years after diagnosis and the 5-year survival rates for these cancers are about 20% and 60%, respectively[13,14]. Conversely, in prostate cancer relapses occur late, with over 90% of the patients still alive 15 years after initial diagnosis[15]. In breast cancer, relapses occur with a peculiar bimodal distribution: a first peak appears generally 1-2 years and a second peak 4-5 years after surgery, followed by a tailed extension up to 15 years[16,17].
BREAST CANCER SUBTYPES AND ADJUVANT THERAPIES
Breast cancer is the most common cancer diagnosed among women. In spite of improved management over the past 30 years, it remains the leading cause of cancer-related mortality for women world-wide[18]. Therapy and prognosis are largely determined by the biological and molecular characteristics of the primary tumor and its size and spreading at time of diagnosis[19,20]. There are three main clinically relevant biological subtypes: Oestrogen/Progesterone receptor positive breast cancer (ER+/PR+), HER2 amplified (HER2+) breast cancer and Triple Negative Breast Cancer (TNBC; i.e., ER-, PR-, HER2-)[21-23]. Based on gene expression signatures four main molecular subtypes have been reported: Luminal A and B, HER2+, and basal-like, which overlap largely, but not fully with the ER+/PR+; HER2+and TNBC biological subtypes, respectively[24,25]. Both biological and molecular classifications have prognostic and predictive (therapeutic) relevance[19,20,24,25]: ER+tumors are treated with adjuvant anti-estrogen therapies (e.g., tamoxifen) while HER2+tumors are treated with HER2 inhibitors (e.g., trastuzumab), in addition to radiotherapy and chemotherapy, if necessary[19,26]. TNBC, has no molecular target useful for targeted therapy yet and adjuvant radio- and chemotherapies are still the standards of care[22,27]. The rational for administering adjuvant therapy after surgery is to eradicate disseminated tumor cells (DTC) or micro-metastases to decrease the risk of relapse. A large body of evidence from adjuvant studies suggest that ER+breast cancer benefits less from chemotherapy compared to ER-breast cancer[28]. This is particularly true for the luminal A molecular subtype of ER+breast cancer, which has a low rate of proliferation. Luminal A tumors have lower rates of pathologic complete response to chemotherapy compared to the highly proliferative ER+luminal B breast cancer subset, as demonstrated with neo-adjuvant anthracycline/taxane-based chemotherapy[29,30]. For HER2+breast cancer the introduction of HER2 inhibitors as adjuvant therapy, in combination with taxanebased chemotherapy, has vastly decreased the risk of metastatic progression and greatly improved survival in this cancer subtype[23,31]. TNBC is highly proliferative and respond better to chemotherapy compared to ER+cancers[22,32]. Today no established targeted therapy exists for TNBC[33]. Interestingly, a fraction of TNBC are rich in tumor infiltrating lymphocytes and this infiltration has been associated with improved diseasefree survival and overall survival OS, suggesting that immune cells may contribute to therapy success and implying the possibility of applying check-point inhibitors-based immunotherapies for these patients[10,22].
In spite of an approx 30%-35% decrease in mortality over the past 35 years due to combined systematic early detection and improved adjuvant therapies, there are still about 20%-25% of breast cancer patients, all stages combined, that will eventually succumb to their disease due to formation of therapy resistant metastases. This corresponds to about 95,000 and 40,000 women in Europe[1](EU 28) and the United States[2], dying every year, respectively[18,19,34]. As of today, there are no effective, curative therapies for metastatic disease. Therapy-resistance and therapy-related toxicity limit therapeutic options[35].
METASTATIC DISSEMINATION
Cancer metastases is a multistage process. Cancer cells have to first escape from the primary tumor, survive in the circulation as circulating tumor cells (CTC), seed at distant sites as DTC and grow to colonize the new tissue and form secondary tumors[36-38]. Growing evidence indicates that metastases are formed by a subset of tumor cells with “stem cell-like” features[39-41]that also associated with resistance to treatments and dormancy[42-44]. Accordingly, molecules controlling stem cell maintenance and differentiation[10,22,45]have been implicated in metastasis, including Wnts, BMPs, TGFb family members, Notch, CD44 and integrins[46,47]. Cancer stem cells (CSCs), in contrast to normal adult stem cells, seem able to revert the hierarchy so that a differentiated cancer cell can revert and recover the stem-like features, while normal, differentiated somatic cells are not able to do so. Thus, CSCs may be rather defined by function than lineage and may represent a form of adaptation of cancer cells to cellular or microenvironmental stress[48-50]. This plasticity may be one reason why by eliminating CSCs as proposed as a new therapeutic approach to eradicate cancer, may actually not be as effective as anticipated[51-54]. Acquisition of CSCs traits has been associated with Epithelialto-Mesenchymal Transition (EMT), a condition endowing cancer cells with increased migratory, invasive, metastatic and therapy resistance capacities[53,55,56]. For example, breast cancer cells undergoing EMT acquire a cell surface phenotype (i.e., CD44high/CD24low) associated with CSCs properties[57]. Accordingly, EMT is a reversible process, as cells that disseminated through EMT and lost epithelial features, can revert back to their epithelial phenotype through an opposite process called Mesenchymal-to-Epithelial Transition (MET)[8]. Both CSCs and EMT are features that are heavily influenced by the microenvironment such as the vascular niches or inflammation (See below).
The genetic and epigenetic basis of metastasis is still not fully elucidated[58]. A current paradigm relies on the notion that the accumulation and selection of genetic mutations and epigenetic alterations is the basis of clonal evolution at the primary site and metastatic dissemination is its ultimate expression[59]. This notion is supported by the clinical observation that primary tumor size is a main risk factor for metastasis. This suggests that metastasis formation occurs rather in late disease stage as the end product of an evolutionary processes in the primary tumor (linear model of metastasis)[8,37,60,61]. According to this model many driver mutations found in the primary tumor are present at the metastatic site, and only a few additional mutations accumulate between primary tumor and metastases[62-64], including in breast cancer[65-68]. In the other hand, comparative genomic hybridization analysis in breast and other cancers revealed that DTCs display significantly more genetic aberration than in the primary tumors[69-72]. These observations imply that metastatic cells disseminate early during tumor development and then progress independently from the primary tumor through multiple steps of genetic mutations. Therefore, the parallel progression model of metastasis has been proposed[60]. Importantly, the two models are not mutually exclusive: a first vague of cancer cells may disseminate early during tumor formation, for example at the time of oncogene activation or EMT induction[73-75], followed by the late dissemination of cells that acquired metastatic properties through local evolution[64,76]. Recently, evidence for the parallel progression model in breast cancer was reported by using experimental models. By studying metastasis in a HER2-driven murine model of breast cancer, Harperet al.[77]showed that cancer cells migrate away from early lesions shortly after HER2 activation. In this model over 80% of the metastases were derived from early disseminated cancer cells. Using the MMTV-HER2 breast tumor model, Harperet al.[77]identified a subpopulation of ERBB2+/p-p38low/p-ATFlow/TWIST1high/E-CADlowearly cancer cells that are invasive and can spread to distant organs (early disseminated cancer cells - eDCC). By using intra-vital imaging they showed that ErbB2+eDCC precursors invaded locally, intravasated and lodged in target organs through a WNT-dependent EMTlike dissemination program. Strikingly, although the majority of eDCCs were TWIST1high/E-CADlowand dormant, they eventually formed metastasis. This experimental observation supports the notion that DTC/DCC can remain dormant for prolonged periods before resuming growth to form macroscopic metastases. As current adjuvant treatment in breast cancer seems to have reached a plateau in term of survival benefits, understanding how DTC and micro-metastases adapt to the distant environment, survive and eventually resume growth to form macroscopic metastases may identify new therapeutic opportunities[1,8,78].
In both the linear and parallel tumor progression models, the genomic instability of tumor cells is the basis of the evolutive process. The variability emerging among tumor cells within the same tumor tissue is referred to as intratumoral heterogeneity and is also found within metastases[79]. This heterogeneity may also be responsible to the presence of tumor cells with low and high tumorigenic potentials, the latter being CSCs or cancer initiating cells (CICs)[80]. Moreover, it is believed that heterogeneity also exists inside the CSC population itself. In such a scenario, metastasis progression and resistance to anti-tumor treatment are thought to be due to clonal evolution and selection much alike Darwinian evolution[59]. Intratumoral heterogeneity may also contribute to tumor dormancy or escape from it. Marusyket al.[81]have used the MDA-MB-468 tumor cell modelin vivoto show that a IL-11 expressing tumor population is able to drive non-cell-autonomous tumor growth from dormant tumor cells. This indicates that re-establishment of certain heterogeneity is necessary for tumor growth after seeding or treatment. In line with this observation, Acetoet al.[82]have reported that, CTC clusters have higher ability to seed metastasis compared to single CTC, involving at least in part altered DNA methylation[83]. Furthermore, Kmieciaket al.[84]showed that the heterogeneity of breast cancer cells in the levels of IFN-γ receptor α expression could determine a selective dormancy. Tumor cells expressing high levels of IFN-γ receptor α are eliminated by CD8+T cells, while tumor cells with low expression levels do not die and remain dormant in the presence of IFN-γ producing CD8+T cells. Thus, tumor heterogeneity contributes to tumor dormancy by providing cells with different genetic and biological features.
METASTATIC BREAST CANCER DORMANCY
As mentioned before, breast cancer metastasis occurs with a bimodal distribution with two peaks: a first one 1-2 years and a second one at 4-5 years after surgery, followed by a tailed extension up to 15 years[17,85,86]. These observations are inconsistent with a model of continuous growth kinetics and rather suggestive of discontinuous growth, thereby implying a period of dormancy[16,17,85-87]. Clinical observations also revealed that timing of appearance of metastasis has a similar profile for the different breast cancer subtypes, suggesting that after primary tumor removal, DTC from distinct subsets evolve following similar patterns but with different dominances (i.e., TNBC and HER2+cancers tend to relapse at the early peak, compared to ER+/PR+cancer which tend to relapse at the second peak or later[85,86,88,89]. These observations also suggest that relapses occurring at peaks may follow inducible and reproducible patterns based on defined mechanisms, for example tumor surgery[90], while relapses occurring in between or in the tailed extension may be due to unpredictable or random events, such as genetic mutations, epigenetic modifications or unrelated inflammatory events[65,67,68,91,92]. Accordingly, these clinical observations were modelled mathematically by considering known basic hazard rates and unknown variables[93,94]. Dormant disseminated cancer cells and micrometastases have been reported in breast cancer patients[95]and in experimental animal models[77,96-98]. Although dormancy is a common phenomenon in breast cancer, the underlying biological mechanisms remain ill characterized. Dormancy might be functionally considered as a transitional, metastable step of cell adaptation to a novel external stress, in particular a “foreign” soil. DTC lacking survival capabilities would rapidly die, while those that have acquired the latter would immediately grow to form macroscopic metastases without latency [Figure 1]. Mechanistically, three forms of cancer dormancy have been described and many genes and molecules involved identified[99-102]: cellular dormancy, microenvironmental (angiogenic) dormancy and immunological dormancy. These three forms of dormancy are not mutually exclusive and it is likely that clinical dormancy is owed by their combination and interrelation.
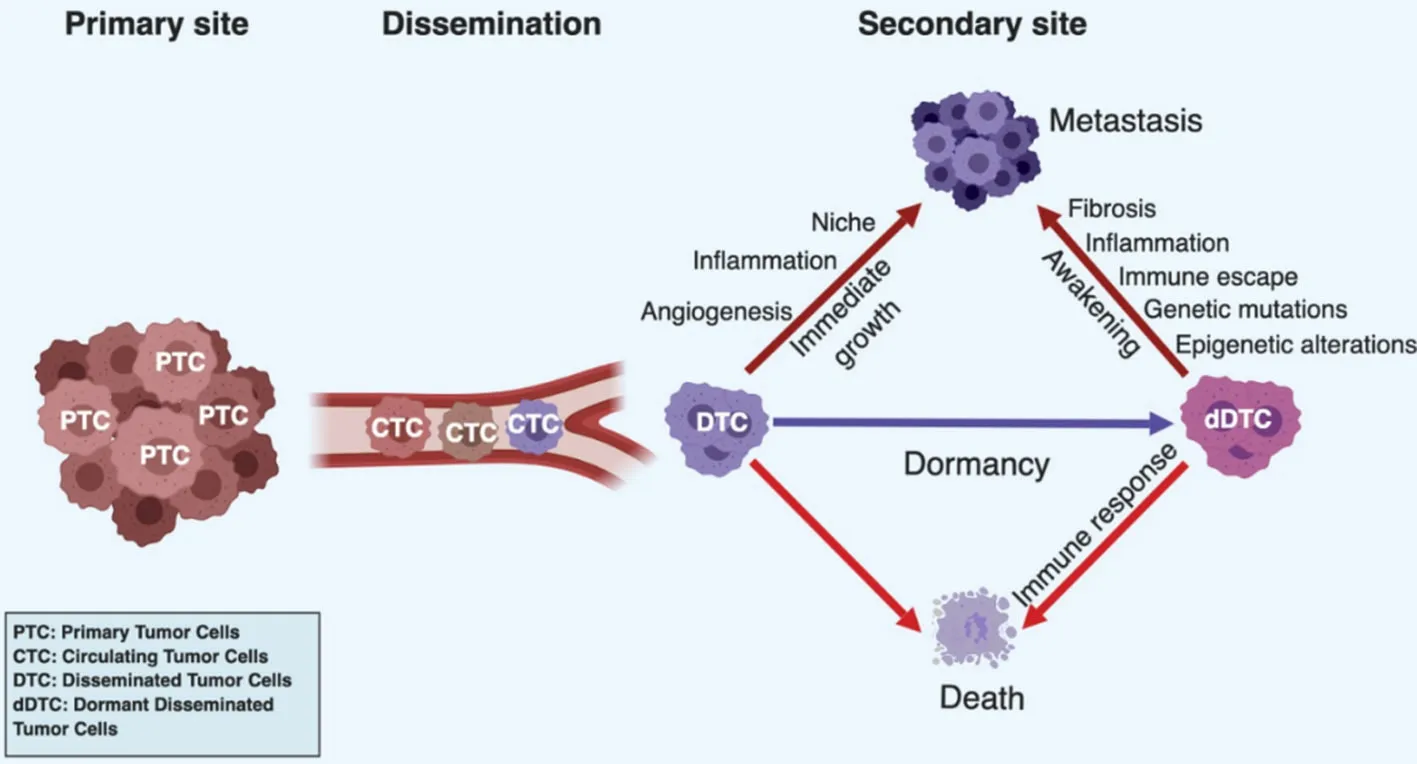
Figure 1. Dormancy in cancer progression. Tumor cells can leave the primary tumor site and enter the systemic circulation as circulating tumor cells (CTCs). Once surviving CTCs have reached a target organ, they seed into a new tissue as disseminated tumor cells (DTCs). Their fate is diverse depending on their cell autonomous capacities and complementary cues provided by the local environment. DTCs can rapidly die if they fail to adapt to the new condition or are killed by the immune system. They can immediately resume proliferation if they have acquired full autonomy for cell survival and proliferation or the local microenvironment provide missing complementary cues. In addition, proliferating cells have to evade the immune system. Alternatively, DTC or small tumor cell clusters, can enter a state of dormancy if cell autonomous or microenvironmental signals are sufficient to maintain survival but do not effectively support growth or the immune system keeps them in check by preventing their expansion. Dormant tumor cells can eventually die by exhaustion, be killed by the immune system or resume proliferation and generate clinically relevant metastases at later time points
CELLULAR DORMANCY: SURVIVAL OF NON-PROLIFERATING SOLITARY CELLS
Dormant DTC cells have developed mechanisms of survival in a foreign environment, but not yet those allowing unrestricted growth. They enter a state of cell cycle arrest (i.e., G0-G1) and survive as nonproliferating solitary cells or as small cell clusters. Accordingly, solitary dormant cancer cells should be negative for the proliferation marker Ki67 as well as for apoptosis markers such as the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). Entry into dormancy and subsequent reactivation seem to be regulated by intrinsic programs and by contextual cues, similar to those involved in the physiological regulation of adult tissue stem cells[103]. Lack of signaling from the matrix seems to play a role in this form of dormancy, as loss of α5β1 integrin expression or function and inhibition of uPAR induce dormancy through the inhibition of the RAS-RAF-ERK signaling pathway, activation of p38/JNK signaling, and induction of p53/Rb-dependent cell-cycle arrest[103-105]. By studying DTC in an experimental model of head and neck squamous cell carcinoma, Sosaet al.[106]have shown that epigenetic upregulation of orphan nuclear receptor NR2F1 (COUP-TF1) plays a critical role in maintaining DTCs in a dormant state. This finding has been further extended by a study in breast cancer patients, in which NR2F1 was tested as a dormancy marker. Breast cancer patients with < 1% NR2F1highDTC in bone marrow aspirate had all systemic relapse within 12 months, while only half of the patients with > 50% NR2F1highremained metastasis-free[107].
Alterations in cell signaling have been found associated with tumor dormancy. High levels of ERK1/2 activity lean toward a higher proliferation, so the ratio of ERK1/2 to p38 MAPK regulates the cell cycle suggesting that the cross talk between mitogenic and stress signals may be relevant to induce cellular dormancy[108]. Impinging on the PI3K signaling cascade was shown to lead to quiescence and the activation of autophagy[109]. Dormant tumor cells express high levels of ARHI, an inhibitor of the PI3K-AKT cascade, and ARHI silencing awakens dormant cells of several tumor types, and promotes their proliferation[110,111]. Consistently, very low or absent AKT signaling in DTC from breast cancer patient has been shown to correlate with the state of dormancy[112]. Strikingly, however, mTOR, a known target of AKT, is found to be activated in quiescent cancer cells. This activation is independent of AKT and is maintained by the small GTPase RHEB with anti-apoptotic activity and itself under control of the stress-regulated transcription factor ATF6α and high p38 kinase activity. Thus, mTOR seems to be a critical node integrating diverse signaling pathways regulating dormancy[111,113,114]. Recently Malladiet al.[115]have shown that human breast and lung carcinoma cells express stem-cell like SOX transcription factors, which induces autocrine expression of DKK1, a WNT inhibitor, resulting in a state of metastatic latency and immune evasion consistent with dormancy.
Interaction with the extracellular matrix is also implicated in controlling dormancy[116], paralleling the role of cell-matrix interaction in physiological CSC niches[117]. For example, the uPAR interaction with a5b1 integrin increases ERK activity and integrin binding to fibronectin fibrils suppresses p38 activity, increases ERK activity and promotes cell proliferation. Accordingly, low uPAR-expressing cells that are growth arrested (dormant)in vivo, have a high p38/ERK activity ratio and fail to assemble fibronectin fibrils and ligate a5b1 integrin[104]. Integrin a5b1 was shown to promote survival of growth-arrested breast cancer cells reminiscent of breast cancer dormancy in bone marrow[118]. Collagen-rich matrix (fibrosis) at the metastatic site is also a critical determinant of DTC transition from dormancy to metastatic growth[116]. For instance, hepatocellular carcinoma cells that colonize rigid matrix resume growth and proliferation through TGF-β1 signaling, while cells colonizing a softer matrix remain dormant[116]. The proliferative switch of dormant DTCs in response to fibrosis in a mouse model of breast cancer was shown to be also mediated by β1 integrin[116]. 3D-in vitro models and furtherin vivostudies demonstrated the critical role of type I collagen in the proliferative fate of DTCs[119-121]. Another matrix protein, periostin, produced by TGF-β1-stimulated stromal fibroblasts and angiogenic blood vessels[47,122], can drive DTCs escape from dormancy through WNT signaling in breast carcinoma[123]. Interfering with extracellular matrix-integrin interaction, in particular b1 integrins, has been proposed as therapeutic approach to promote dormancy, including in breast cancer[124,125].
Autophagy appears to promote DTCs survival and dormancy by maintaining DTCs viable under conditions of microenvironmental stress[126]. Autophagy also promotes survival of DTC against chemotherapy-induced cell stress[126]. On the other side, in breast cancer the lack of autophagy is associated with early tumor recurrence and escape from dormancy[127]. It is important to note, that cellular dormancy is not just a feature of cancer cells but it also occurs in normal cells. For example, hair follicle (bulge) stem cells, muscle stem cells (satellite cells), hematopoietic and liver stem cells are rather quiescent under homeostatic conditions and can be rapidly activated during tissue regeneration and repair[128-130]. These observations raised the idea that cancer cells may hijack the signaling cascade used physiologically by these cells to enter, maintain and exit dormancy during homeostatic and regenerative conditions[103,131].
SENESCENCE, A SPECIAL FORM OF CELLULAR DORMANCY?
The term senescence was originally introduced to describe primary human fibroblasts in culture, that after reaching a maximal number of passages (cell divisions) entered a state of permanent growth arrest[132,133]. Senescent cells remain metabolically and synthetically active but show significant alterations in morphology[132-134]. Following these original observations, scientists discovered that replicative senescence is driven by telomere shortening[135]. Telomerase, an enzyme reconstituting telomers, is expressed in germ line cells, early embryonic cells, but not in normal cells, and is re-expressed in most cancer cells[134]. Besides telomere shortening, other stimuli have been found to induce the senescence[136]including DNA damage[137], chemotherapy[138]and oncogene activation[139]. Despite the nature of the stimulus initiating the senescence cascade, the signals ultimately converge to the p53/p21 and/or p16INK4a/pRB pathways[136]. The tumor suppressor proteins DEC1 (Deleted in Esophageal Cancer) and Decoy Receptor 2 are also used as senescence markers in some cell types[140]. Importantly, when oncogenic HRAS (HRAS12V) was used to transform immortalized embryo fibroblasts into tumorigenic cells[141], unexpectedly, it induced senescence in normal cells associated with the accumulation of p53 and p16INK4a[142]. Thus, senescence may act as a tumor-suppressor mechanism in response to oncogene activation[133]. Generally, overexpression or sustained activation of one of the tumor suppressors p53, p21, p16INK4a, or pRB is sufficient to induce senescence[136,143-145]. Importantly, growth arrest caused by senescence is considered as irreversible as senescent cell could not be stimulated to resume proliferation by exposure to growth factors[136,141,143]. However, the genetic or epigenetic alternations which cause the shift of the senescence maintaining mechanisms, such as the inactivation of tumor suppressor genes p53 and/or p16INK4a, could potentially push the cells to re-enter the cell cycle[136,143].
It is not clear whether senescence is one of the mechanisms that drives tumor dormancy and late relapse, but there are some potential links suggesting so. Senescence-associated secretory phenotype (SASP) defines the spectrum of factors secreted by senescent cells. It consists of a mixture of chemokines, cytokines, growth factors and proteases, many of which are pro-inflammatory[136]. The cytokine GM-CSF (granulocytemacrophage colony-stimulating factor, also known as CSF2), one of the components from SASP, induces differentiation of myeloid dendritic cells, which present tumor-associated antigens (TAAs), resulting in the activation of the immune system, enhanced immunosurveillance and improved tumor control[146,147]. Furthermore, Braumülleret al.[148]showed that adaptive TH1 cell are capable, via the combined secretion of IFN-γ and TNFR to induce tumor cells senescence. In conclusion, while senesce is a mechanism capable of negatively controlling tumor growth, its relevance in dormancy is not fully demonstrated.
MICROENVIRONMENTAL DORMANCY: DEFICIENT SUPPORT FROM THE MICROENVIRONMENT
In this form of dormancy, the fate of the DTCs is mainly controlled by the immediate, and possibly distant, host environment[126,149]: cancer cells proliferate but fail to grow as a tumor mass because proliferation is balanced by cell death[99]. This situation has been described first when disseminated cancer cells fail to induce blood vessels[150]. During the early stage of tumor spreading, DTCs associate to preexisting (coopted) blood vessels where oxygen and nutrient levels are highest[151]. Importantly, quiescent mature blood vessels keep DTCs in a state of dormancy through angiocrine communication[122]. With the growing tumor mass, however, the increasing metabolic demand call for the formation of novel blood vessels through an angiogenic switch[152]. Several molecules, including hypoxia-inducible factor 1, vascular endothelial growth factor (VEGF), placental growth factor and angiopoietin-1, are induced upon metabolic stress or hypoxia and initiate endothelial cell sprouting from preexisting vessels[153]. Angiogenic endothelial sprouts secrete periostin and TGF-β1 to create a microenvironment that promotes DTCs exit from dormancy and accelerates proliferation[122]. Thus, the failure of initiating the angiogenic switch will keep the tumor mass small, a condition called angiogenic dormancy[154]. Conversely, a short-term angiogenic burst may awaken dormant tumor cells[155]. For example, in the mouse model of Lewis lung carcinoma, dormant micrometastatic cells can be induced to to grow by the expression of VEGF and the recruitment of bonemarrow-derived endothelial progenitor cells[156].
Importantly, inflammatory cells recruited to the tumor microenvironment are critical inducer of the angiogenic switch and lack of recruitment may therefore contribute to angiogenic dormancy[11,157]. In particular the polarization of tumor associated macrophages toward the M2 phenotype results in the formation of a metastatic niche favoring tumor cell outgrowth[158]. In addition, the angiogenic endothelium triggers a T helper 2-mediated inflammatory response, which can accelerate metastatic outgrowth in tumor models[159]. The hemopoietic stem cell niche also supports quiescence and survival through the CXCL12/CXCR4 pathway and Src pathway[47,160]as well as the TGF-β2-rich bone marrow microenvironment[161]. On the other hand, expression of VCAM1 on DTCs promotes escape from dormancy. This is due to the recruitment of osteoclast progenitors via α4β1 integrin binding to VECAM1 causing the breakdown of the bone matrix and stimulation of DTC to grow and form metastases[162,163]. Likewise, metastatic outgrowth following skeletal traumas was associated with bone remodeling in the reactivation of DTCs via TNFα, IL1β, IL6 and prostaglandin E2 production[163]. Thus, the cellular tumor microenvironment, in particular inflammatory cells, endothelial cells and osteoclasts should be considered as critical regulators of tumor dormancy.
IMMUNOLOGICAL DORMANCY: ACTIVE CONTROL BY THE IMMUNE SYSTEM
The immune system can specifically recognize tumor cells through TAAs and initiate an anti-tumor immune response through a multi-step process called immunoediting[164,165]: initially, the immune response can effectively eliminate cancer cells and no metastases are formed (elimination phase). If a balance between the tumor suppressive immune response and tumor evasion is achieved (equilibrium phase), the tumor mass remains constant resulting in a state of immunological dormancy[166]. Tumor cells may eventually succeed in evading the immune system through a combination of genetic evolution and exhaustion of the immune system, and resuming growth to form macroscopic metastases (escape phase). Evasion involves multiple mechanisms, including the down-regulation of TAAs or MHC molecules, secretion of immunosuppressive cytokines, (e.g., IL-10 or TGF-b), recruitment of regulatory T cells (Treg), myeloid derived suppressor cells (MDSC) or alternatively (M2) - polarized macrophages or by expressing immunosuppressive molecules on the cell surface, such as PDL1 or B7[167-169], in multiple cancer types, including breast cancer[170]. The occurrence and relevance of immunological dormancy in human cancer is difficult to grasp, as the absence of biomarkers of dormancy makes it hard to investigate in patients. Also, there is paucity of clinically relevantin vivomodel for experimental studies[100]. Pommieret al.[171]have recently reported that patients and mice with pancreatic ductal adenocarcinoma contained single quiescent DTCs lacking MHC-I expression, which enabled them to escape immunity and establish latent metastases. Four cases of breast cancer transmission to immunosuppressed transplant recipients from a single, clinically healthy donor have been recently described[172]. The latency time to metastasis formation ranged from 16 months to 6 years, and transmissions did not occur in a patient that discontinued immunosuppression. This unintentional situation suggested that donor tissues harbored DTCs, which persisted in a latent state because of immune control, while the immunosuppression of the recipient allowed the DTCs to resume growth[172]. Once cells have escaped immunosurveillance it is unlikely that they can re-enter a second state of immunological dormancy, because the mechanisms that eluded immunosurveillance are generally part of the genetic or epigenetic tumor clonal evolution and therefore irreversible[59,91].
The recent observation that immunosuppressive immune cells promote angiogenesis[157], and that some angiogenic factors, including VEGF, are immunosuppressive, established an important cross-talk between these two systems[173-175]. They implicate that suppression of angiogenesis may stimulate the immune system and that reversal of immunosuppression may inhibit angiogenesis in a bi-directional cross-talk. In support of this notion, several trials have been performed to improve the anti-tumor immune response with antiangiogenic drugs[176]. Combination of anti-angiogenic agents with immunotherapy is currently being considered as strategy to improve the response rates and duration of immunotherapy[177]. For example, VEGF has been shown to suppress the immune-response by impinging on maturation of dendritic cells[178,179]. Thus, inhibition of VEGF abrogates its immunosuppressive effect and improves the antitumor effect of adoptive cellular immunotherapy[180]. Combining anti-VEGF therapies with check-point inhibitors (e.g., anti-PD-L1) has shown synergy and positive outcomes in phases I to III studies, particularly in patients with high VEGF levels[177]. Using the 4T1 experimental model of TNBC we have demonstrated that inhibition of tumor angiogenesis with the anti-VEGFR-2 antibody DC101, attenuated the inhibitory effect of MDSC on T cell proliferation and decreased the frequency of Tregs in primary tumors and lung metastases[181]. Combined angiopoietin-2 and VEGF inhibition was shown to promote superior vascular regression, tumor necrosis, and enhanced the perivascular recruitment of cytotoxic T lymphocytes, as compared to the single agents in multiple cancer models. Addition of a PD-1 blocker unleashed the cytotoxic activity resulting in improved tumor control[182,183]. These findings support the rationale for combining anti-angiogenic therapy with immune checkpoints inhibitors in cancer therapy, including in breast cancer. In addition to immunosuppressive angiogenic molecules, endothelial cells themselves also play a direct role in modulating cellular dormancy. Quiescent endothelial-derived thrombospondin-1 induces sustained quiescence of DTC in breast cancer, while sprouting vessels wake dormant DTC and promote their outgrowth[122,184,185][Figure 2].
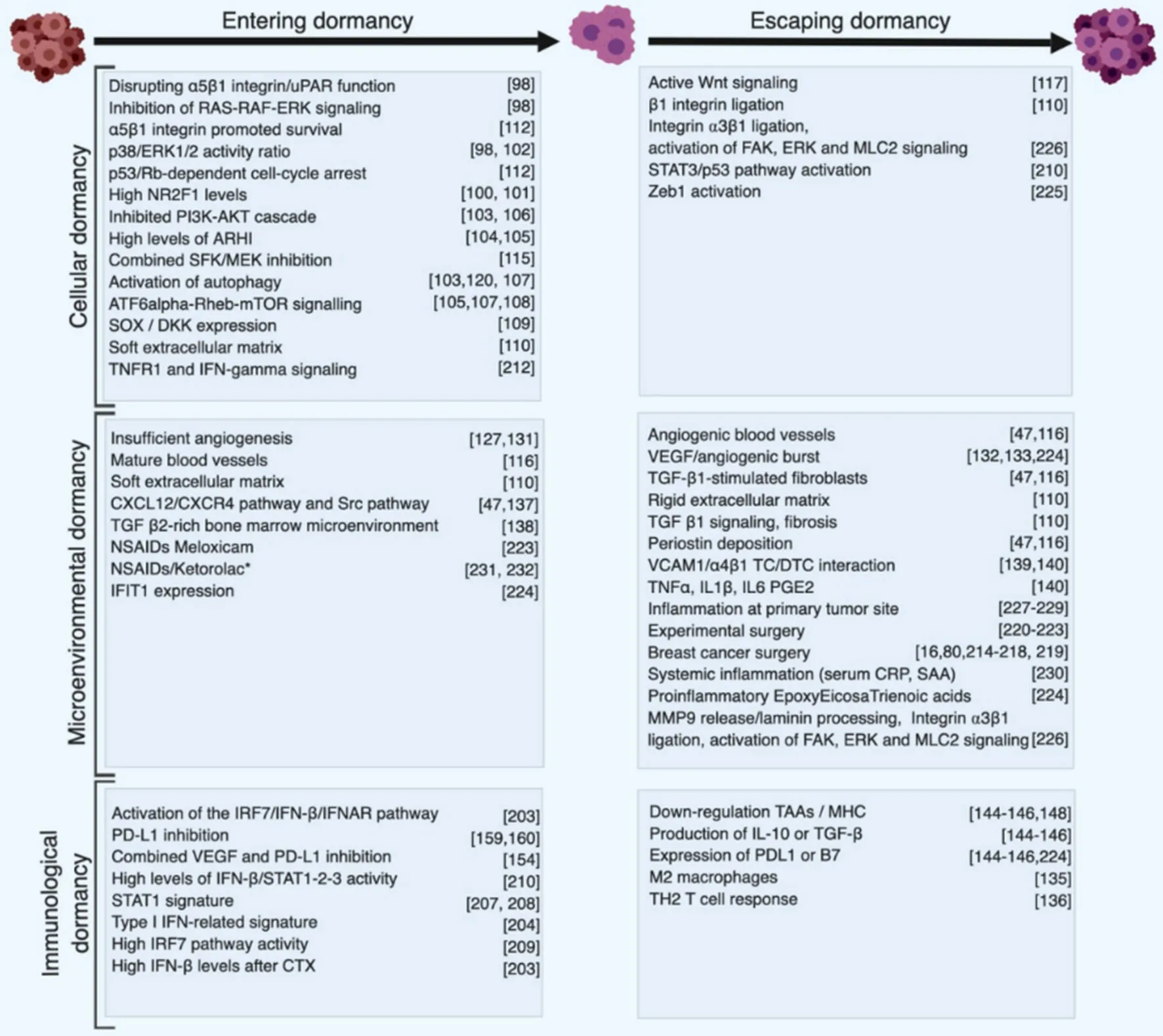
Figure 2. Synthetic summary of cellular and molecular events and interventions associated with dormancy. Cellular and molecular events promoting entering into dormancy or escape from dormancy are listed based on their implication in the three forms of dormancy. Classification is based on the main mechanisms and does not consider potential cross talk between different types of dormancy
DORMANT CANCER CELLS ARE RESISTANT TO THE CYTOTOXIC EFFECTS OF CHEMOTHERAPY
Adjuvant chemotherapy significantly improves breast cancer patient survival, in particular for TNBC, by decreasing the hazard of relapse after surgical removal of the primary tumor[26,32,186-188]. Relapses and metastases however still occur in a fraction of patients and are likely due to tumor cells that had already invaded the surrounding tissue, lymphatic vessels and lymph nodes, or disseminated through the blood stream to the bone marrow and distant sites prior to surgery and resume grow at a later time point[60,77,189]. As it is generally assumed that adjuvant chemotherapy acts by killing tumor cells, a corollary of this assumption is that relapses are due to DTC resuming growth at a later time point after surviving chemotherapy. Dormant, poorly proliferative DTC may not be affected by chemotherapy and may persist upon treatment since chemotherapeutic drugs mainly target highly proliferating cells. This has been shown for instance in acute lymphoblastic leukemia, colorectal, lung, liver and breast cancers[190-196]. In addition, chemotherapy pre-exposed DTC may develop mechanisms of chemoresistance and become less responsive to subsequent chemotherapies, as is often observed at relapse in patients. CSCs have been shown to be chemo-resistant and to be responsible for post-therapy relapses[197]. Chemotherapy causes enrichment of CSCs thereby facilitating recurrences and resistance to further chemotherapies in multiple cancers including glioma, ovarian, liver, colon, breast cancers[197]. Resistance involves multiple mechanisms, such as activation of signaling pathways prominent in stem cells (e.g., WNT, NOTCH, HEDGEHOG)[198-200], but also pathways that are frequently mutated and activated in cancer, in particular the EGFR-HER2/PI3K/PTEN/Akt/mTORC pathway[198,200,201]. CSC are often enriched at sites of chronic hypoxia leading to the activation of the HIF pathway[200,202]. Interestingly, HIF activation leads to the initiation of protective pathways, including WNT and NOTCH, and genes of the ATP-binding cassette (ABC) drug transporter family members, such as MDR1, MRP1 and ABCG2 which are responsible for the eラux of cytotoxic drugs from the cells[203-208]. Additional, HIFindependent mechanisms have been reported[209]. In short, the mechanisms behind the long-term beneficial effects of adjuvant chemotherapy remain in part elusive and cannot be fully explained by the direct cytotoxic activity of chemotherapy as dormant/CSC that are mostly responsible for late relapses that are highly resistant to chemotherapy.
IMMUNE RESPONSE AND CHEMOTHERAPY IN BREAST CANCER
Cumulating evidence indicates that tumor infiltrating lymphocytes (TILs) play an active role in controlling progression and clinical outcome in breast cancer, particularly in highly proliferative TNBC and HER2+cancers[210-212], and in conjunction with chemotherapy[213,214]. This is particularly relevant to TNBC, as these cancers present the richest presence of TILs, most notably CD8+T lymphocytes, and tertiary lymphoid structures[211,215,216]. Increased numbers of infiltrating TIL in TNBC, are associated with an improved pathological complete response to chemotherapy[217], decreased rates of recurrences and improved survival[210,216,218]. Evaluation of TILs in breast cancer has been recommended as an immunological biomarker with prognostic and potentially predictive values, mainly in TNBC and HER2-amplified breast cancers[219]. In TNBC, expression of antigen presenting MHC class II pathway molecules is associated with a better outcome, consistent with the hypothesis that a functional antigen presentation pathway may trigger a protective antitumor immune response in response to chemotherapy[220]. Ladoireet al.[221]demonstrated that pathological complete response to neoadjuvant chemotherapy of breast carcinoma resulted in the disappearance of tumor-infiltrating FOXP3+regulatory T cells and the increase in tumor infiltrating cytotoxic TiA1+and granzyme B+T cells, consistent with the induction of an antitumor immune response by chemotherapy. While the association between lymphocytic infiltrates, and improved outcome after chemotherapy appears to be strongest in TNBC and HER2+breast cancer subtypes, the association with luminal tumors is less clear, and may be limited by the reduced immune infiltration or by the greater tumor heterogeneity of these tumors[222]. Several recent experimental studies have shown that chemotherapy can induce a therapeutic anti-tumor immune response. For example, Maet al.[223,224], reported that anthracyclinebased chemotherapy induces the release of ATP by dying breast cancer cells, which promotes the recruitment and differentiation of CD11c+CD11b+Ly6Chighantigen presenting cells. Depletion or preventing tumor infiltration by these cells abolished the anti-tumor immune responses elicited by anthracyclines[223,224].
Besides these desired immunological effects, chemotherapy can also induce unwanted, immune-mediated tumor-promoting effects. For example, increased expression of TNFa in the breast tumor microenvironment due to chemotherapy, induces CXCL1/2 expression via NF-κB activation in breast cancer cells and initiates a paracrine loop involving myeloid cell-derived S100A8/9 to enhance cancer cell survival and chemo-resistance. Inhibition of CXCR2 blunt this TNFa-induced response and augments the efficacy of chemotherapy, particularly against breast cancer metastasis[225].
Taken together, there is growing evidence to support the notion that chemotherapy, in particular those based on anthracyclines, can elicit an effective anti-tumor immune response in breast cancer, mainly in the TNBC and HER2+subtypes.
CHEMOTHERAPY-INDUCED IMMUNOLOGICAL BREAST CANCER DORMANCY
One important question emerging from these studies is whether the cytotoxic activity of chemotherapy and the elicited immune response, dominated by CD8+T lymphocytes, may be sufficient to effectively kill cancer cells or whether additional mechanisms may be involved. This is particularly relevant to dormant DTC as these cells are naturally less responsive to chemotherapy due to the fact that they are not or low proliferative. We were interested in the possibility of whether a short chemotherapy treatment, induce a long-lasting immune response leading to immunological dormancy. We recently addressed this question experimentally[226]. To this end, we treated the TNBC-like 4T1 cells with high dose Methotrexate and Doxorubicinin vitroand characterized thein vitroandin vivobehavior of the surviving cells (MR20 and DR500 derived from Methotrexate and Doxorubicin treatment, respectively). The hallmark of the surviving cell lines was the dormant phenotype at the primary (MR20 in the mammary gland) or at the metastatic (MR20 and DR500 in the lung) site. MR20 cells grew significantly slowerin vitrocompared to parental 4T1 cells and this was due to increased cell death, while there was no significant alteration in the cell cycle. When injected orthotopically in the mammary fat pad of immunocompetent BALB/c mice, only about half of the MR20-injected mice developed tumors with a longer latency (between 6 week and 4 months) compared to parental 4T1 cells (between 2 and 4 weeks). These tumor cells grew with nearly the same rate as parental 4T1 tumors and were highly metastatic. DR500 cells formed primary tumors but no lung metastases. Both conditions were consistent with dormancy. In BALB/c mice MR20 and DR500 cells twisted the immune response from CD11b+Gr1+MDSC-dominated to CD4/8 T cell, B cell and DC dominated response. When injectedin vivoin immunodeficient NSG mice, however, treated cells formed tumors without latency. We then performed a genome wide gene expression analysis of parental 4T1, MR20 and DR500 cells and the dominant trait observed was a type I IFN gene expression signature and the sustained activation of the IRF7/IFN-β/IFNAR pathway. Upregulated IRF7 expression in treated cancer cells was responsible for suppressed mobilization of CD11b+Gr1+MDSCs, increased expansion of DCs, T and B lymphocytes and chemo resistance. Silencing IRF7 or blocking IFNAR1 resulted in escape from dormancy in vivo. Elevated levels of IFN-β were present in the blood of mice injected with dormant cells, while MR20 cells escaping dormancy and forming late tumors no longer expressed IFN-β. Interestingly, the dormant D2.0R murine breast cancer cells that are generally considered as a model of cellular dormancy, also induced a T-cell twisted immune response and had a constitutive active IRF7/IFN-β/IFNAR pathway [Figure 3]. To collect evidence that the activation of the type I IFN pathway was associated with a better response to chemotherapy in patients, we monitored IFN-β levels in serum samples of ER-breast cancer patients treated with neoadjuvant chemotherapy (TOP trial NCT00162812). Patients with detectable IFN-β during neoadjuvant therapy had significantly longer distant metastasis free survival (DMFS) compared to patients with undetectable levels[226].
TYPE I IFN RESPONSE TO CHEMOTHERAPY MEDIATES IMMUNOLOGICAL DORMANCY IN BREAST CANCER
Taken together our results demonstrate that sustained activation of the IFN-β/IFNAR/IRF7 signaling axis in chemotherapy-treated TNBC-like murine breast cancer cells instigates immunological dormancy. Elevated levels of IFN-β in the serum of TNBC patients during adjuvant therapy correlates with a shorter DMFS. Acute exposure of breast cancer cells to chemotherapy induced IFN-β expression[226]. Thus IFN-β may be considered as a potential therapeutic tool to improve chemotherapy efficacy and clinical outcome as well as a potential predictive biomarker to identify respondingvs. non-responding patients. While the implication of type I IFN in dormancy is novel, previous reports have implicated it in acute anti-tumor response to chemotherapy. Sistiguet al.[227]reported that anthracycline-based chemotherapy rapidly induced the production of type I IFN in cancer cells via TLR3 activation resulting in CXCL10 secretion promoting an anti-tumor immune response mimicking those induced by viruses. Moreover, a type I IFN-related signature predicted clinical responses to anthracycline-based chemotherapy in breast cancer patients. Using a panel of ER-breast cancer PDXs before and after chemotherapy, Legrieret al.[228]demonstrated activation of the IFN/STAT1 pathway and expression of IFN-inducible genes, early after chemotherapy treatment. IFN-a deficient DC were shown to accumulate in aggressive breast cancers favoring the expansion of Tregs implicating that IFN-α deficiency may contribute to tumor immune tolerance and poor clinical outcome[229]. Previous studies based on tumor-derived IFN signatures have shown that IFN-regulated genes may correlate with favorable outcomes. A STAT1 signature before therapy was associated with a better response to neoadjuvant chemotherapy[230]and better prognosis in TNBC and HER2+breast cancers[231]. High IRF7 pathway activity in human breast cancer predicted bone relapse-free survival and, and protected mice against bone metastasis[232]. In the same study, treatment with IFN-α improved bone metastasis-free survival[232]. High level of IFN-β activates STAT1, STAT2 and STAT3 to facilitate cell autonomous cellular dormancy of melanoma repopulating cells[233]. Taken together, our recent data extend the role of type I IFN in immunoediting in cancer[234]to a putative role in inducing immunological dormancy after chemotherapy in TNBC. An important outstanding question raised by our study, concerns the effector mechanism of T cell mediated dormancy. While it is likely that direct CD8+T cell mediated killing and immune control is involved, particularly since CD4+Th1 T cells produce IFN-γ which upregulates MHC expression on tumor cells thereby facilitating killing by CD8+T cells[235], it is plausible that additional mechanisms may be involved. One possibility is suppression of tumor angiogenesis by the secretion of anti-angiogenic factors, such as CXCL9 and CXCL10 by CD4+T cells[235]. Another putative mechanism is induction of senescence. CD4+Th1 T cells also produce TNF-α, which in combination with IFN-γ induces an irreversible state of cellular senescence keeping DTC dormant[235,236]. Ongoing projects in our lab are aimed at unraveling the effectors steps.
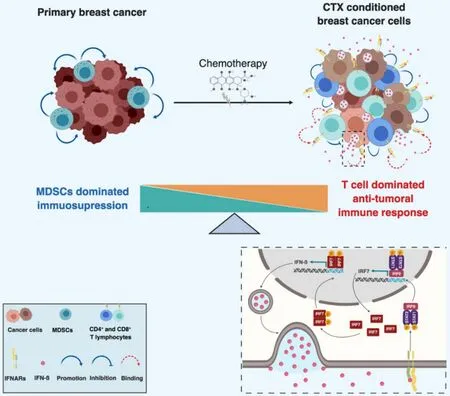
Figure 3. Schematic model of chemotherapy-induced immunological dormancy. Primary breast cancer cells escape immune elimination by inducing the expansion of myeloid derived suppressive cells (MDSC) which also promote tumor growth. Chemotherapy induces a type I IFN response in treated tumor cells, resulting in an autocrine and self-sustained increase of IRF7 expression and activation, which in turn induces expression and secretion of IFN-β. Secreted IFN-β binds to IFNAR and induces signaling in both immune cells and tumor cells. IFNARs signaling in tumor cells activates STAT1/STAT2/IRF9 complex which further induces the expression of IFN-β responsive genes including IRF7 resulting in a sustained autocrine IFN-β expression and secretion. Paracrine activation of IFNARs on immune cells stimulates the expansion of tumor suppressive lymphocytes (e.g., CD4+ and CD8+ T cells) and prevents the mobilization of MDSCs, resulting in the switch of the immune response from immunosuppressive to anti-tumoral
BREAKING DORMANCY: IMPACT OF INFLAMMATION
Clinical and experimental observations indicate that dormant tumor cells can resume proliferation and generate macroscopic metastases at various times after primary tumor treatment[17,85]. Escape from dormancy could be initiated by cell autonomous events, such as oncogenic mutation or inactivation of tumor suppressor genes. Alternatively, alterations in the host micro- or macro-environment may promote escape from dormancy. Metastatic growth following surgical removal of the primary tumor itself is often observed in clinical settings with predictable patterns, including in breast cancer[16,86,237-241]. Reconstructive surgery after mastectomy for breast cancer significantly accelerates relapse rates proportionally to the extent of surgery, when compared to primary surgery. However, no worsening in long-term survival was reported, consistent with an effect on breaking dormancy and accelerating progression but not altering the overall outcome[242]. Enhancement of metastatic growth induced by experimental surgery has been long observed in animal models, suggesting that surgical wounding itself may be directly involved in breaking dormancy[243-245]. Recently Krallet al.[246]described an experimental model that links the wound-healing response after surgery to the outgrowth of DTC at distant sites. The link is mediated by the systemic inflammatory response induced after surgery suppressing a tumor-specific T cell response, thereby resulting in tumor growth. Consistent with these findings, perioperative anti-inflammatory treatment significantly reduced tumor outgrowth. Epoxyeicosatrienoic acids, a family of pro-inflammatory molecules, stimulated escape from dormancy in several tumor models independently of the primary tumor and was associated with increased production of VEGF by endothelial cells[247]. Recently, it was reported that lung inflammation promotes escape from tumor latency by inducing ZEB1 expression, a regulator of the EMT[248]. Another recent study reported that neutrophil extracellular traps (NET) produced by neutrophils during repeated acute inflammation awaken dormant cancer cells in a mouse model of breast cancer[249]. The releasing of neutrophil elastase and MMP9 from NET then remodel laminin in the extracellular matrix rendering it accessible to a3β1 integrin. Ligated a3β1 integrin leads to the activation of FAK, ERK and MLC2 signaling resulting in the awakening of the dormant cancer cells[249].
Clinical and experimental evidence suggests that sustained inflammation may also promote relapses[91]. Correlations between inflammation at primary tumor site and risk or recurrences were reported for several cancers including oral[250], endometrial[251]and breast cancers[250,252]. Elevated levels of circulating C-reactive protein and serum amyloid A, two proteins of the inflammatory response, are associated with reduced overall survival in breast cancer, independently of body mass index, age and tumor type and stage, consistent with inflammation being involved in breaking dormancy[253]. Interestingly, perioperative administration of the analgesic nonsteroidal anti-inflammatory drug (NSAID) ketorolac was reported to suppress early breast cancer relapse particularly in TNBC patients[254]and to reduce distant recurrences in patients with increased BMI[255]. Taken together, there is compelling clinical and experimental evidence indicating that inflammation promotes breast (and other) cancer relapses by breaking dormancy.
OUTLOOK AND PERSPECTIVES
Tumor dormancy is widely accepted as one discrete step during multistage tumor progression and bears considerable therapeutic potential[256]. The rapid translation of this innovative concept to the patient is limited by the paucity of therapeutic tools currently available in the clinic. Treatments directly aimed at DTC by critical cell survival and proliferation pathways (e.g., PI3K-AKT or MAPK pathways), stem cell pathways (e.g., WNT, NOTCH) or cell adhesion molecules (e.g., b1 integrin) would be virtually excluded for such an approach given their expected long-term systemic toxicities. We are proposing here selected strategies based on limiting the host (unwanted) inflammatory response and stimulating the (wanted) anti-tumoral-immune response that may be rapidly tested in clinical-translation studies in breast cancer [Figure 4].
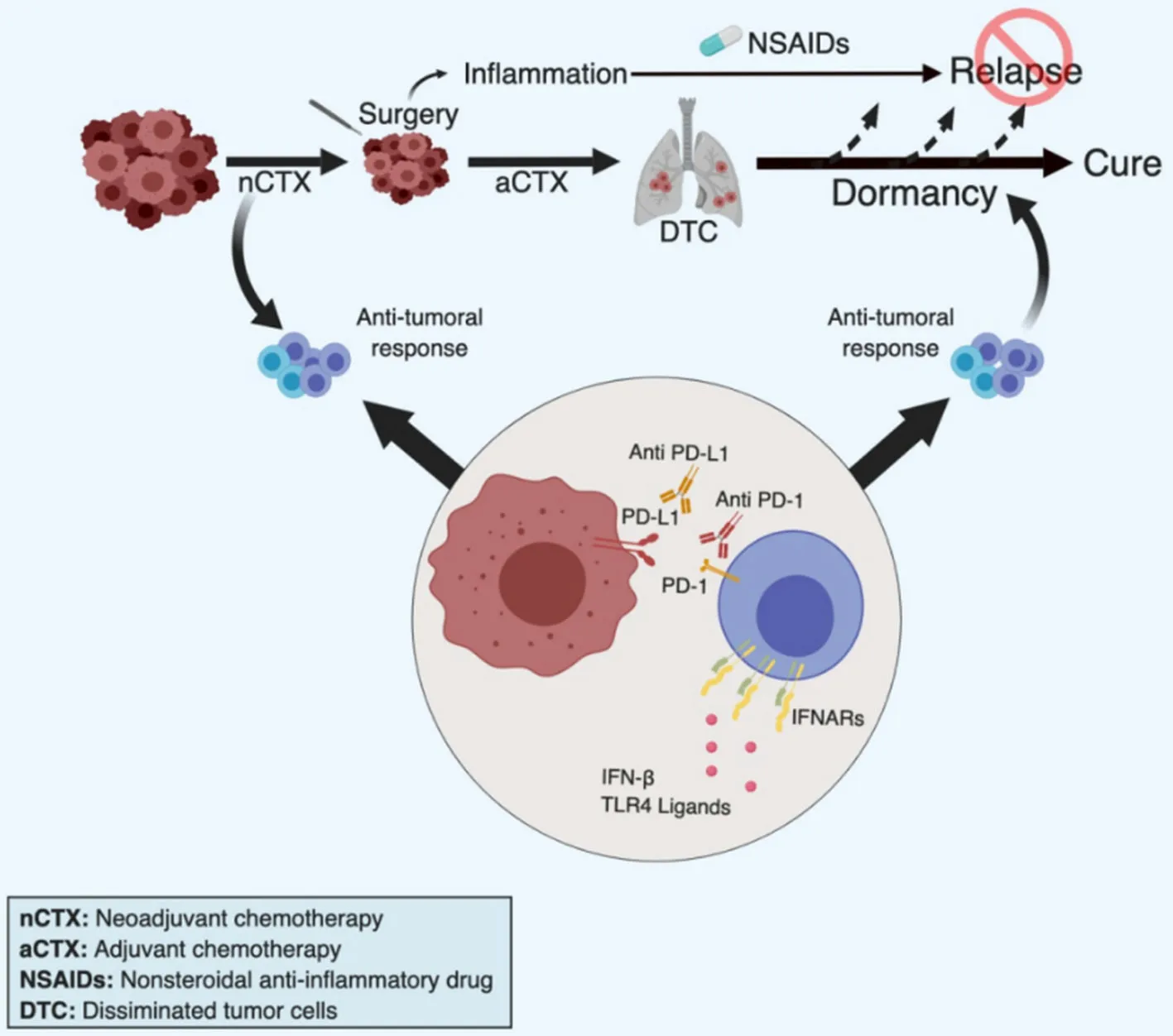
Figure 4. Strategies to improve chemotherapy-induced immunological dormancy. Based on work by others and us, we propose four clinically feasible approaches to induce or maintain breast cancer dormancy, primarily in TNBC. Firstly, we propose neo-adjuvant chemotherapy (nCTX) to promote chemotherapy-induced immune response. Chemotherapy may be pursued as adjuvant therapy (aCTX) if necessary. Secondly, during and following surgery we propose the administration of NSAIDs (in particular Ketorolac) to prevent surgery-associated inflammation that may potentially promote relapses. Thirdly, the cytotoxic immune response could be stimulated by providing type I IFN, or inducers of an interferon response such as TLR4 ligands, in particularly in low IFN-producing patients. Fourthly, addition of checkpoint inhibitors, such as anti-PD1/PDL-1 antibodies, may be applied to maintain the immune response active
NSAIDs
There is growing clinical and experimental evidence that inflammation can trigger cancer relapse, in particular in breast cancer, and that NSAIDs treatment can prolong dormancy and delay or reduce relapses[246,248,249,252,254,255,257]. The overall positive safety profile of aspirin and other NSAIDs would make them realistic candidate drugs for such long-term therapies[258]. Even more interesting, as a short perioperative treatment with ketorolac has been shown to significantly decrease the risk of breast cancer relapses particularly in obese patients[255], a short term NSAID treatment at time of surgery may have long-lasting effects through suppression of surgery-associated inflammation.
Neoadjuvant chemotherapy
A second approach to consider, is to exploit the ability of chemotherapy to elicit an effective immune response in breast cancer, particularly in lymphocyte-infiltrated TNBC or HER2+tumors[211,212,222,224,226,227,259].
Our and other’s results indicate that chemotherapy induces an effective anti-tumor immune response upon tumor cell treatment[214,226,227]. Clinically this implies that neo-adjuvant/pre-operative chemotherapies may be more effective in inducing a long-lasting, protective immune response compared to classical adjuvant/post-operative therapies, due to the larger targeted tumor mass[224,260]. Indeed, neoadjuvant chemotherapy is already used in highly proliferative breast cancers (i.e., Luminal B, HER2+and TNBC) with high frequencies of pathological complete responses (pCR). Interestingly, paclitaxel, in combination with trastuzumab, induced a high rate of pCR in HER2+patients, likely due to the synergy between the immunomodulating properties of these drugs[260]. Ladoireet al.[221]reported that pCR to breast cancer neoadjuvant chemotherapy was associated with the disappearance of tumor-infiltrating FOXP3+Tregs and recruitment of CD8+T cells, consistent with the induction of an antitumor immune response by chemotherapy.
Type I IFN
In our experimental model we have shown that Type I IFN response is essential to elicit a state of immunological breast cancer dormancy[226]. Others have shown that exogenous addition of type I IFN boosted an insufficient response to chemotherapy in an experimental model of breast cancer and that a type I IFN-related signature predicted clinical responses to anthracycline-based chemotherapy in breast cancer patients[227]. We demonstrated that patients with high levels of serum IFN-β during neoadjuvant therapy have a better outcome compared to patients with low levels[226]. This suggests that administration of type I IFN during neo-adjuvant therapy may be effective in mounting a long-lasting immune response in particular in those patients with low endogenous type I IFN levels. Type I IFN, in particular IFN-α has been already tested as immunostimulatory anti-cancer agent, especially in melanoma and kidney cancers, albeit with mild results, in part also due to the need of repeated administrations and its intrinsic toxicity[261-263]. Alternatively, less toxic inducers of Type I IFN response, such as TLR-ligands of STING stimulators may be considered[227,264].
Check point inhibitors
A complementary strategy to IFN administration could be the use of check point inhibitors, in particular anti-PD-1/PD-L1 antibodies to relieve tumor-induced immunosuppression[265]. In breast cancer, immunotherapy is being explored, in particular in patients with tumors expressing PD-L1 and infiltrated with lymphocytes[266]. Potential response to PD-1 or PD-L1 inhibitors was demonstrated in metastatic TNBC[267]and HER2+breast cancers[268]. However, because the number of potential neoantigens available for immune response in most breast cancers, responses are modest compared with other cancers such as lung and melanoma, the use of check-point inhibitors may require combination with other therapies[269]. Therefore, combination with neo-adjuvant chemotherapy (causing the release of tumor antigens), antiangiogenesis therapies (suppressing inhibitory cues)[182]or Type I IFN (acting immunostimulating)[270]may be more effective and should be explored.
Biomarkers of dormancy
There are currently no specific non-invasive biomarkers to monitor breast cancer dormancy of clinical utility[271]. Such markers would allow personalized follow up and accelerate therapeutic decisions in case of evidence of disease progression. CD44+/CD24-CTC subsets along with combinatorial expression of uPAR and b1 integrin, as well as proliferation and apoptosis markers in CTC of early breast cancer patients, have been explored for potential use as biomarker of dormancy or aggressiveness[272,273]. Genomic analysis (i.e., SNP/CNV) of circulating ctDNA was shown to potentially identify breast cancer patients with dormant/minimal residual disease[274]. Also, serum inflammatory markers might serve as biomarkers of relapse in disease-free patients, as inflammation is associated with escape from dormancy but will likely be unspecific and of limited sensitivity[275]. Serum IFN-β levels were associated with longer DMFS (as a surrogate of dormancy) in our model of chemotherapy-induced dormancy and during neoadjuvant therapy in patients with favorable outcome[226]. Also, we observed a shift in peripheral blood leucocyte populations in our experimental model, from a Gr1+CD11b+cell dominated response in mice with progressive tumors toward a CD4+/CD8+-, B cell and CD11c+cell-dominated response in mice with dormant tumors[226]. Thus, Serum IFN-β levels and immunophenotyping should be explored for their potential use as biomarkers of breast cancer dormancy.
CONCLUSION
Advances in the understanding of the mechanisms of breast cancer dormancy have raised the hope to therapeutically exploit dormancy to prevent relapses and overt metastatic disease. To date many potential therapeutic targets and strategies have been considered and proposed for clinical testing[100,185]. However, many of these approaches would be difficult to apply to patients due to lack of suitable drugs, potential longterm toxicity and over all complexity in the in their clinical translation. Recent reports implicating a T cell based immune response in the therapeutic effects of chemotherapy including dormancy, have opened novel perspectives for a feasible clinical translation. Administration of chemotherapy before tumor removal (i.e., neoadjuvant chemotherapy) may be explored for improved effects on immunological dormancy compared to conventional, post-surgery, adjuvant chemotherapy. Drugs with potential beneficial effects on promoting or prolonging dormancy, such as NSAIDs to suppress inflammation, type I interferons, check-point inhibitors or anti-angiogenic drugs to stimulate the immune response, are available for clinical use and could be tested in combination with chemotherapy. Thus, the observation that chemotherapy can induce a state of immunological dormancy adds a new therapeutic effect to the older class of anti-cancer drugs and opens unanticipated therapeutic opportunities for clinical translation in breast cancer (and possibly other cancers).
DECLARATIONS
Acknowledgments
We apologize for not being able to cite all published work relevant to this topic due to space limitation and selective focus of the article.
Authors’ contributions
Conceiving the paper: Peyvandi S, Lan Q, Lorusso G, Rüegg C
Writing the paper: Peyvandi S, Lan Q, Lorusso G, Rüegg C
Performed the work of immunological dormancy in the laboratory: Peyvandi S, Lan Q, Lorusso G, Rüegg C
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the Swiss National Sciences foundation (31003A_159824/1, 31003A_179248/1); the Swiss Cancer League (KSF3513-08-2014, KSF-4400-02-2018); NCCR Molecular Oncology, NCCR Bio-Inspired materials, the Medic Foundation, the Sassella Stiftung, the 3R foundation, and the European Union under the auspices of the FP7 collaborative project TuMIC (HEALTH-F2-2008-201662).
Conflicts of interest
All authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
The Author(s) 2019.
 Journal of Cancer Metastasis and Treatment2019年5期
Journal of Cancer Metastasis and Treatment2019年5期
- Journal of Cancer Metastasis and Treatment的其它文章
- Operative treatment of metastatic breast cancer in the spine with regard to molecular phenotypes
- Autophagy in breast cancer metastatic dormancy: tumor suppressing or tumor promoting functions?
- The lncRNA BORG: a novel inducer of TNBC metastasis, chemoresistance, and disease recurrence
- Stem cells, immortality, and the evolution of metastatic properties in breast cancer: telomere maintenance mechanisms and metastatic evolution
- Training and evaluation of a knowledge-based model for automated treatment planning of multiple brain metastases
- ESR1 alterations and metastasis in estrogen receptor positive breast cancer