
Stem cells, immortality, and the evolution of metastatic properties in breast cancer: telomere maintenance mechanisms and metastatic evolution
2019-07-29 08:43NathanielRobinsonDerekTaylorWilliamSchiemann
Nathaniel J. Robinson, Derek J. Taylor, William P. Schiemann
1Department of Pathology, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
2Department of Pharmacology, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
3Case Comprehensive Cancer Center, Case Western Reserve University, Cleveland, OH 44106, USA.
Abstract Breast cancer is the most significant cause of cancer-related death in women around the world. The vast majority of breast cancer-associated mortality stems from metastasis, which remains an incurable disease state. Metastasis results from evolution of clones that possess the insidious properties required for dissemination and colonization of distant organs. These clonal populations are descended from breast cancer stem cells (CSCs), which are also responsible for their prolonged maintenance and continued evolution. Telomeres impose a lifespan on cells that can be extended when they are actively elongated, as occurs in CSCs. Thus, changes in telomere structure serve to promote the survival of CSCs and subsequent metastatic evolution. The selection of telomere maintenance mechanism (TMM) has important consequences not only for CSC survival and evolution, but also for their coordination of various signaling pathways that choreograph the metastatic cascade. Targeting the telomere maintenance machinery may therefore provide a boon to the treatment of metastatic breast cancer. Here we review the two major TMMs and the roles they play in the development of stem and metastatic breast cancer cells. We also highlight current and future approaches to targeting these mechanisms in clinical settings to alleviate metastatic breast cancers.
Keywords: Breast cancer, cancer evolution, cancer stem cells, metastasis, telomerase
INTRODUCTION
Breast cancer is the most common malignancy and most frequent cause of cancer-related death in women globally[1]. The vast majority of breast cancer-related morbidity and mortality can be ascribed to metastasis, which occurs in ~30 percent of cases and underlies ~90 percent of breast cancer deaths[2,3]. Metastasis is a multistage cascade that commences when cancer cells migrate from their primary tumor of origin and undergo hematogenous dissemination that terminates in the seeding and colonization of distant organs[4]. This so-called “invasion-metastasis cascade” serves as an evolutionary bottleneck that requires disseminated tumor cells (DTCs) to: (1) activate migratory and invasive programs; (2) survive within the vasculature in an anchorage- independent manner; (3) interact with other circulating cells to facilitate survival and extravasation; and (4) coordinate tissue-specific signaling inputs to persist in unfamiliar microenvironments[5-7]. Thus, metastasis can be viewed as a process of clonal selection whereby a heterogeneous primary tumor gives rise to subpopulations that are fit to traverse the invasion-metastasis cascade. Following tissue colonization, these disseminated subclones retain growth-permissive features of the original primary tumor and undergo further evolution and clonal expansion within metastatic microenvironments[8,9].
Metastatic evolution occurs via a number of distinct yet spatiotemporally overlapping mechanisms, including linear and parallel progression of monophyletic or polyphyletic founder clones[10]. Cancer stem cells (CSCs) are fundamental components of tumors that enable the maintenance of emergent clonal populations yielded by evolutionary forces[11-13]. CSCs are operationally defined by their self-renewal and tumor-initiating capacities; that is, a single CSC can recapitulate a tumor in its entirety, including a stable CSC pool[14]. Historically, stochastic clonal evolution was believed to be mutually exclusive with a tumor developmental hierarchy built upon a stem cell population[15,16]. More recent evidence suggests that there is a relationship between tumor evolution and CSCs that manifests through at least two mechanisms. First, the CSC population itself becomes highly heterogeneous during tumor development, indicating that CSCs are directly subjected to selective pressures[17,18]. Second, non-stem cancer cells that define unique genetic and epigenetic lineages can be reprogrammed into CSCs[19,20]. Thus, the plasticity that exists within and between stem and non-stem cancer cells provides a bidirectional route to engender clones that harbor distinctive properties, including the ability to metastasize. Of note, the functional significance of CSC evolution in the development and progression of multiple malignancies has been extensively documented[21-23].
Numerous pathways that exert control over the metastatic propensity of cancer cells do so by regulating the production or function of CSCs. For instance, Wnt/β-catenin signaling in both the primary tumor and metastatic microenvironments enhances breast CSC self-renewal and metastatic colonization[24,25]. Likewise, inhibiting Wnt signaling abrogates metastatic outgrowth by depleting the CSC population[26,27]. Similarly, vascular endothelial growth factor (VEGF) activates stem programs in breast cancer cells via VEGF receptor (VEGFR)-and neuropilin (NRP)-dependent cascades[28,29]. VEGF can additionally push breast CSCs to undergo endothelial-like differentiation, thereby promoting tumor vascularization and cancer cell dissemination[30]. The NF-κB transcription factor pathway also acts as a critical regulator of breast CSC function[31]. In particular, microenvironmental stimuli from resident stromal cells, extracellular matrix components, and the local immune milieu activate NF-κB signaling to sustain CSC development[25,32,33]. As a result, NF-κB inhibitors demonstrate potent activity against breast CSCs[34]. Related to these events, CSC expansion is associated with the epithelial-mesenchymal transition (EMT), a process whereby epithelial cells lose their intrinsic polarity and markers of differentiation and adopt features of mesenchymal cells, including enhanced migration and invasiveness[35,36]. Key transcription factors that orchestrate EMT in breast cancer, such as Snail, Slug, and Twist1, simultaneously play a role in the acquisition of stem-like traits[37]. Importantly, both Wnt/β-catenin and NF-κB signaling exert direct transcriptional control over these EMT-associated factors[36,38]. Furthermore, EMT induces upregulation of VEGF, which bolsters the activities of β-catenin and NF-κB and promotes angiogenesis to support CSC self-renewal and permit dissemination[39-41]. In short, breast CSC survival and maturation are determined by a confluence of cell-intrinsic and microenvironmentderived signals that are transduced through parallel EMT-dependent and -independent circuits.
CSCs, like embryonic and tissue stem cells, possess replicative immortality[42], a process achieved in part by activating telomere maintenance mechanisms (TMMs)[43,44]. As outlined below, TMMs function within a network that unites cellular immortalization with processes, including EMT, that drive the development and outgrowth of metastatic cells. Telomeres, therefore, serve as essential mediators of CSC maintenance and consequent metastatic evolution. In addition, the results detailed below implicate telomere homeostasis as an attractive target for novel therapeutics to treat metastatic breast cancer.
TELOMERES AND TELOMERE DYNAMICS IN CSCS AND METASTATIC CELLS
Telomeres are nucleoprotein complexes located at the ends of linear chromosomes that safeguard against chromosomal instability and the loss of genetic information during cell division[45]. In humans, the DNA component of telomeres is composed of tandem (TTAGGG)nrepeats with a 3' single-stranded overhang that invades telomeric duplex DNA to form a protective loop[46]. These DNA regions are coated with proteins that collectively constitute the shelterin complex. Shelterin proteins serve to shield telomeres from illicit activation of DNA damage responses (DDRs); they also maintain genome integrity and recruit factors responsible for regulating telomere length[47,48]. In somatic (i.e., non-immortalized) cells, telomeres shorten during iterative rounds of cell division. To combat this event, stem cells and cancer cells maintain their telomeres using one of two TMMs: telomerase or alternative lengthening of telomeres (ALT). Telomerase is a reverse transcriptase enzyme composed of an RNA moiety (TERC, also known as TR) that provides a template for telomeric DNA synthesis and a protein moiety (TERT) that facilitates telomerase recruitment and carries out its polymerase activity[49]. In contrast, ALT relies upon homology-directed, recombination-dependent synthesis of nascent telomeric DNA[50]. ALT requires transient deprotection of telomeres coupled to activation of a DDR that is accompanied by telomere extension in a manner similar to break-induced DNA synthesis[51,52]. DDR activation occurs in response to alterations in telomeric and subtelomeric chromatin structure that are brought about by loss of the chromatin remodelers ATRX and DAXX[53,54]. Notably, evidence of each of these mechanisms has been found in breast cancer and can be correlated with specific histologic subtypes or disease stages[55,56]. These findings support the idea that TMM identity may impact breast cancer progression, including the onset of metastasis.
While TMM acquisition has been identified as a feature of both stem and non-stem cancer cells, these processes play an essential role in preferentially sustaining the CSC population[42]. By virtue of their replicative immortality, CSCs function as progenitors that exist over a sufficient timescale for evolution to take place. Remarkably, telomere shortening appears to be a primary driving force underlying tumor evolution. Telomere shortening precedes TMM activation[57], which allows for the formation of critically short telomeres that cannot be adequately capped by shelterin. Cells interpret these short telomeres as free DNA ends, which are temporarily repaired by chromosome end-to-end fusions that ultimately induce breakage-fusion-bridge (BFB) cycles[58,59]. BFB cycling leads to complex genomic rearrangements including deletions, non-reciprocal translocations, and formation of dicentric or circular chromosomes[60]. Telomere catastrophe may also yield chromosomal instability that is resolved via chromothripsis or other forms of chromoanagenesis, an event termed telomere crisis[61,62]. Breast cancer-initiating cells can harbor both short telomeres and telomerase activity[41], consistent with the model that telomere shortening instigates genomic instability and CSC evolution while telomere elongation maintains emergent CSC subpopulations [Figure 1] Evidence identifying ALT in breast CSCs has not yet been found. However, ALT has a stem cell origin[63], while ALT activity has been observed in non-breast CSCs[44,63,64]. Future studies examining TMMs in breast CSCs and their connection to genome architecture and tumor heterogeneity will be of great value.
Telomere maintenance proteins have been heavily implicated in many of the central signaling pathways in metastasis[65][Figure 1]. For instance, TERT is capable of regulating Wnt target genes by forming a transcriptional co-activation complex with β-catenin[66]. In addition, TERT directly regulates NF-κBdependent gene expression by binding to the NF-κB p65 subunit at the promoters of target genes[67,68]. Each of these pathways exerts reciprocal control over TERT[69,70], thereby preserving TMM identity and CSC phenotype. Of note, TERT can also bind to the VEGF promoter to stimulate VEGF expression and neoangiogenesis[71]. Other transcriptional regulators of TERT, such as c-Myc, further serve to induce EMT and stemness in breast cancer cells[72-74]. Compared to telomerase, ALT is less well-characterized at a molecular level; therefore, our understanding of its role in EMT and breast cancer metastasis is presently incomplete. Nevertheless, ALT is most often associated with tumors of mesenchymal origin[75], indicating a possible role for ALT in EMT. Accordingly, carcinoma cells exhibiting telomerase dysfunction were driven to adopt a mesenchymal stem-like phenotype, which was accompanied by activation of ALT and the formation of metastatic tumors[76,77]. In breast cancer cells, TERT expression is mutually exclusive with the mesenchymal state[78]. Similarly to TERT, the expression of ALT- associated proteins, such as the Bloom syndrome protein (BLM), is governed by signaling pathways, such as Notch, that are responsible for CSC fate specification and self-renewal[79,80]. Given these findings, it is paramount that future studies explore the significance of the relationship between stemness and telomere plasticity in breast cancer progression.
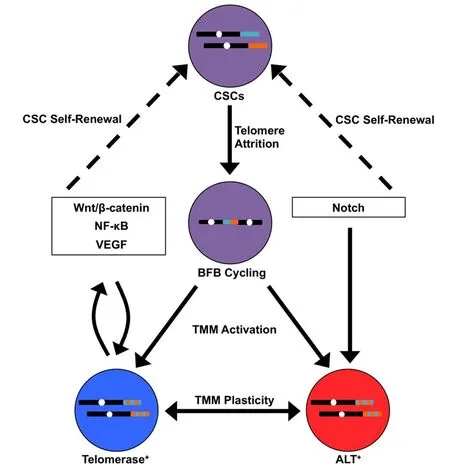
Figure 1. Telomere-centric model of breast cancer stem cell (CSC) biogenesis and metastatic evolution. CSCs (purple) harboring telomeres of a given length (shown for two different chromosomes in teal and orange) undergo telomere attrition as a by-product of self-renewal. This ultimately yields critically short telomeres that are temporarily repaired by chromosome end-to-end fusions, resulting in breakage-fusion-bridge (BFB) cycling (represented by dicentric chromosome). BFB cycling or chromoanagenesis (not shown) cause widespread chromosomal instability (represented by dual-colored telomeres) and the acquisition of new genetic features, including those that are advantageous for metastasis. At the same time, telomere maintenance mechanisms (TMMs) are activated in these new clonal populations, which are defined in part by their reliance on telomerase (blue) or ALT (red). In addition, TMMs exhibit a degree of plasticity, such that TMM identity may interconvert between telomerase and ALT. TMM selection is influenced by signaling pathways that simultaneously promote CSC propagation (dashed arrows). In turn, telomere maintenance proteins directly regulate these signaling pathways, establishing reciprocal feedback loops that coordinate TMM activation and CSC maintenance
TELOMERE-DIRECTED THERAPIES FOR METASTATIC BREAST CANCER: CURRENT AND FUTURE PERSPECTIVES
The functions of telomerase in tumorigenesis have been rigorously interrogated over the last several decades, as has the potential to target telomerase therapeutically[65,81]. The telomerase inhibitors BIBR1532 and GRN163L (also known as Imetelstat) display high efficacy in depleting the CSC pool and disrupting breast cancer metastasis[82-85]. Indeed, Imetelstat was assessed in a Phase I clinical trial for recurrent or metastatic breast cancer, although the trial was suspended due to dose-limiting toxicity[81]. In addition to such toxicity concerns, the success of telomerase inhibitors in clinical trials has thus far been moderated by the inherent complexity of telomere homeostasis. First, telomere shortening-induced senescence can be bypassed in the absence of functional p53 or other components of the DDR machinery[86]. Second, the critically short telomeres and chromosomal instability associated with telomere crisis are disproportionately associated with metastasis[87,88]. Thus, the evolution of DTCs that underlie metastatic disease may be enhanced unwittingly by therapies that promote telomere shortening. Despite these challenges, telomerase remains an appealing therapeutic objective in need of innovative targeting approaches in which these evolutionary considerations are taken into account.
Emerging telomerase-targeting strategies include cytotoxic small molecules that act as substrates for telomerase as well as anti-telomerase immunotherapies[89-92]. Current immunotherapeutic platforms are primarily centered on telomerase peptide or dendritic cell vaccines, which can be engineered to elicit either CD4+ or CD8+ T cell antitumor responses[93]. These strategies are being assessed in diverse preclinical settings, including breast cancer. Indeed, the telomerase peptide vaccine Vx-001 is progressing through clinical trials for advanced solid tumors[90]. More recent investigations have examined the feasibility of adoptive transfer of anti-telomerase chimeric antigen receptor (CAR) T cells for treating triple-negative breast cancer[94]. Future studies into the generalizability of anti-telomerase CAR T cell therapy to other breast cancer subtypes, as well as the efficacy of these diverse immunotherapeutic approaches in clinical settings will be of tremendous value.
Although the functions of specific ALT-associated proteins have been elucidated, their utility as therapeutic targets for ALT-driven cancers has only recently been investigated. For example, the DNA damageresponsive kinase ataxia-telangectasia and Rad3-related (ATR) is activated secondary to depletion of ATRX, which leads to persistent retention of replication protein A (RPA) at telomeres and generation of a recombinogenic substrate. Inhibition of ATR, in turn, triggers apoptosis of ALT-positive cells[95]. BLM, a RecQ DNA helicase, unwinds telomeric G-quadruplex structures and coordinates 5'→3' end resection during telomere recombination[96,97]. Accordingly, a recently-developed small molecule inhibitor of BLM may possess great potential as an anticancer agent against ALT-driven tumors[98]. Finally, topoisomerase IIIα (Topo IIIα) associates with BLM and regulates the topology of telomeric recombination intermediates. Interestingly, genetic inactivation of Topo IIIα selectively reduces the survival of ALT-positive compared to telomerase-positive cells[99]. Moreover, telomerase activity is enhanced in the surviving fraction of Topo IIIα-deficient cells[100], suggesting that telomerase activation provides a pathway for chemoresistance. Thus, targeting TMMs may best be achieved using a multidrug regimen consisting of multiple anti-TMM agents or an anti-TMM agent in combination with chemotherapy or other targeted agents[101]. The effectiveness of these therapeutic modalities in eliminating breast CSCs and in treating metastatic breast cancers remain intriguing and important open questions.
CONCLUSION
By overseeing multiple pathways that promote breast cancer stemness, EMT, and metastasis, telomeres function as critical nodes in the nexus between cellular immortalization, tumor evolution, and disease progression. The selection of TMM likely exhibits a high degree of plasticity in different tumor cell types or across disparate stages of breast cancer development, including metastasis. Indeed, TMM selection may itself be subject to evolutionary dynamic forces. In addition, the plasticity inherent in TMM identity has farreaching prognostic and therapeutic implications. Tumors driven by distinct TMMs may show sensitivity or resistance to specific treatments, which has substantial impact on patient survival. Moreover, different subpopulations within a single tumor (e.g., stem vs. non-stem cells) may be reliant upon unique TMMs. Such TMM heterogeneity may beget residual, resistant clones that underlie disease recurrence. In the future, gaining a deeper understanding of telomeres and the pathways controlled by the telomere machinery will provide immense insight into the origin, progression, and eradication of one of the world’s deadliest cancers.
DECLARATIONS
Acknowledgments
Members of the Schiemann Laboratory are thanked for critical comments and reading of the manuscript.
Authors’ contributions
Conception and study design: Robinson NJ, Schiemann WP
Drafted and revised the manuscript: Robinson NJ, Taylor DJ, Schiemann WP
Availability of data and materials
Not applicable.
Financial support and sponsorship
Research support was provided in part by the National Institutes of Health (CA236273) to Schiemann WP, (CA186571) to Taylor DJ; and (T32 GM007250 and F30 CA213892) to Robinson NJ. Additional support was graciously provided by the METAvivor Foundation (Schiemann WP), and by pilot funding from the Case Comprehensive Cancer Center’s Research Innovation Fund, which is supported by the Case Council and Friends of the Case Comprehensive Cancer Center (Schiemann WP), and from the Case Clinical & Translational Science Collaborative (Schiemann WP). Finally, Taylor DJ is also supported by the American Cancer Society (RSG-13-211-01-DMC).
Conflicts of interest
All authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
 Journal of Cancer Metastasis and Treatment2019年5期
Journal of Cancer Metastasis and Treatment2019年5期
- Journal of Cancer Metastasis and Treatment的其它文章
- The moIecuIar interaction of ADAMTS-1 and fibuIin-1 and its potentiaI contribution to breast cancer bioIogy
- ESR1 alterations and metastasis in estrogen receptor positive breast cancer
- Training and evaluation of a knowledge-based model for automated treatment planning of multiple brain metastases
- The lncRNA BORG: a novel inducer of TNBC metastasis, chemoresistance, and disease recurrence
- Autophagy in breast cancer metastatic dormancy: tumor suppressing or tumor promoting functions?
- Chemotherapy-induced immunological breast cancer dormancy: a new function for old drugs?