
Autophagy in breast cancer metastatic dormancy: tumor suppressing or tumor promoting functions?
2019-07-29 08:43:50AlyssaLaBelleFlynnWilliamSchiemann
Alyssa La Belle Flynn, William P. Schiemann
1Department of Pharmacology, Case Western Reserve University, Cleveland, OH 44106, USA.
2Case Comprehensive Cancer Center, Case Western Reserve University, Cleveland, OH 44106, USA.
Abstract Breast cancer is the second leading cause of cancer-associated death in women in the United States, with more than 90% of those deaths attributed to metastasis. Breast cancer metastasis is incurable and possesses few treatment options and a poor overall prognosis due in part to confounding metastatic attributes, particularly the acquisition of dormancy-associated phenotypes. Dormant disseminated tumor cells can persist for years-to-decades before recurring as highly aggressive, secondary lesions. Dormancy-associated phenotypes are exhibited by breast cancer stem cells (BCSCs), which undergo tumor initiation and unlimited self-renewal. In addition to their specialized abilities to circumvent chemotherapeutic insults, BCSCs also upregulate autophagy during metastatic dormancy as a means to survive in nutrient poor conditions and environmental stress. As such, therapeutic targeting of autophagy is actively being pursued as an attractive strategy to alleviate metastatic disease and the recurrence of dormant BCSCs. Here we review the molecular and cellular features of autophagy, as well as its paradoxical role in both suppressing and promoting mammary tumor development and metastatic progression. Finally, we highlight the clinical challenges associated with therapeutic targeting of autophagy in metastatic breast cancers.
Keywords: Autophagy, breast cancer, cancer stem cells, metastatic dormancy, metastatic relapse
INTRODUCTION
Breast cancer is the second deadliest malignancy in women, accounting for nearly 41,000 deaths in the United States in 2018[1]. More than 90% of the deaths attributed to breast cancer are caused by metastasis, a disease state associated with poor prognosis and little-to-no effective treatment options[2]. Indeed, while initial treatment of breast cancers can be effective and achieve remission, an estimated 30% of lymph node-negative and 70% of lymph node-positive breast cancer patients will eventually relapse 5-20 years following initial diagnosis[3,4]. The period of time between clinical remission and relapse can be attributed to dormancy, a process whereby disseminated tumor cells (DTCs) enter a non-proliferative state coupled with the activation of cellular stress programs[5]. Even in the earliest stages of mammary tumor development, breast cancer cells are actively shed from the growing tumor and traverse the metastatic cascade before colonizing distant metastatic sites[6,7]. These solitary micrometastases can persist in distant organs for years or even decades before emerging as recurrent metastatic tumors. Indeed, experimental evidence andin silicomodeling indicate that dormant DTCs exist in a quiescent state as opposed to one that reflects a balance between cell proliferation and apoptosis[8-12]. Dormant cells upregulate pro-survival factors and are inherently chemoresistant given their non-proliferative state. As such, treatment with currently available therapeutics does little to limit the population of dormant cells in breast cancer patients. In fact, ~62% of breast cancerassociated deaths occur 5 years following diagnosis[13]. As such, the clinical detection and treatment of these recurrent metastases remains challenging due to: (1)difficulties in detecting growing lesions years or decades following remission; (2) limited treatment options that are effective against metastatic disease[14,15]. Despite the fact that systemic relapse following a period metastatic dormancy remains a large unmet clinical burden, the precise mechanism(s) that enable dormant metastatic lesions to reactivate proliferative programs and recur remains incomplete[3]. Here we highlight the importance of breast cancer stem cells (BCSCs) and their reliance upon autophagy to govern the activation and eventual emergence from metastatic dormancy, as well as clinical implications of targeting autophagy therapeutically as a means to alleviate metastatic disease.
BCSCS AND METASTATIC DORMANCY: A ROUTE TO EVADE DETECTION AND THERAPEUTIC ELIMINATION
Recent evidence suggests that DTCs endowed with the ability to survive metastatic dormancy and initiate recurrent metastatic lesions are BCSCs[16-18], which undergo unlimited self-renewal and contribute to tumor initiation[19]. Likewise, genomic analyses of primary and relapsed metastatic breast cancers reveal numerous common driver mutations shared between primary and metastatic tumor lesions in a given patient. As such, these common mutational landscapes implicate the presence of a common malignant cell of origin and support the notion that disseminated BCSCs initiate recurrent metastatic lesions years or decades following clinical remission[20-23]. This process reflects the ability of BCSCs to adopt dormancy-associated phenotypes through several malleable events, including modulation of E-cadherin and lncRNA expression[24,25]. Equally important facets of metastatic relapse are the capacity of BCSCs to evade immune surveillance and resist therapeutic interventions aimed at eradicating residual disease. Amongst the pro-survival strategies activated by BCSCs are: (1) upregulated expression of ATP-binding cassette transporters that mediate cellular eラux of chemotherapeutic agents[26-28]; (2) increased production of Interleukin-4 (IL-4) to suppress apoptosis[29]; (3) enhanced generation of reactive oxygen species in response to radiation[30]; (4) elevated activation of autophagy[16-18,31][Figure 1]. As such, dormant BCSCs are inherently resistant to traditional chemotherapeutic agents and radiation that target rapidly dividing tumor cells. In the succeeding sections, we highlight the role of autophagy in regulating mammary tumorigenesis and dormancy-associated phenotypes during metastatic progression and relapse.
CONTEXT-DEPENDENT ROLE OF AUTOPHAGY IN TUMOR PROGRESSION
Macroautophagy (hereafter referred to as autophagy) is a highly conserved process that maintains cellular homeostasis through the lysosomal degradation of proteins and organelles, a phenomenon that is tightly controlled by autophagy-related genes (ATGs)[32]. The autophagosome cargo protein, p62/sequestosome 1 (SQSTM1), binds to degradation targets and facilitates selective autophagy[33]. Indeed, during the activation of autophagy, ATGs mediate the recycling of p62/SQSTM1-tagged cargo through the formation of doublemembrane vesicles, termed autophagosomes, which fuse with lysosomes to form autophagolysosomes. Lysosomal fusion facilitates the degradation of nonfunctional cellular components and also functions to meet the energy demands of a cell in periods of environmental stress[32,34][Figure 2]. Recent basic and clinical research findings have highlighted the context-dependent role of autophagy in regulating tumorigenesis. Indeed, in the earliest stages of tumor growth and development, autophagy functions as a tumor suppressor, thereby limiting tumor growth. However, once primary tumors or their metastases are established, autophagy can promote tumorigenesis by subverting stress responses, and consequently, facilitating tumor cell survival and disease progression[35][Figure 3]. At present, a thorough understanding of the molecular mechanisms that enable autophagy to both suppresses or promote mammary tumorigenesis is lacking, as are cell- and context-specific signals that underlie the paradoxical functions of autophagy in breast cancers. Future studies need to address these important questions as a means to uncover novel therapeutic strategies aimed at modulating autophagy in patients with metastatic breast cancer.
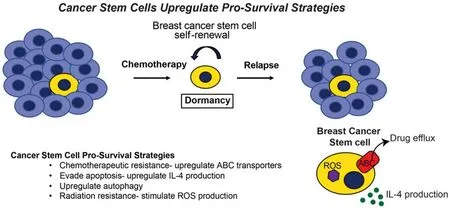
Figure 1. Cancer stem cells upregulate pro-survival strategies. Early in mammary tumor development, breast cancer cells are shed and disseminated from the growing lesion, ultimately colonizing distant metastatic sites before clinical detection of a primary breast tumor. Upon breast cancer diagnosis, neoadjuvant chemotherapy in conjunction with surgical resection, or more traditionally, surgery followed by adjuvant chemotherapy are both effective in eliminating the bulk the primary tumor cells. In contrast to bulk tumor cells, breast cancer stem cells manage to survive chemotherapeutic treatment by upregulating a number of pro-survival strategies, thereby contributing to metastatic relapse following a period of remission and dormancy. In doing so, cancer stem cells can (1) upregulate ABC transporter expression, which evades the cytotoxic activities of chemotherapies; (2) enhance IL-4 production, which inhibits apoptosis; (3) activate autophagy; (4) induce ROS production, which confers resistance to radiation. In addition, breast cancer stem cells also evade apoptosis by lying dormant for years or even decades, a pathophysiological state that further protects these cells from the cytotoxic activities of chemotherapy and radiation, and from the apoptotic activities engendered by metabolic, hypoxic, and environmental stressors
AUTOPHAGY AND TUMOR SUPPRESSION
Anecdotal evidence indicates that autophagy can act as a barrier to prevent tumor initiation in a number of solid tumors, including those of the breast. For instance, autophagy is readily induced by the tumor suppressors PTEN and p53, while their inactivation in developing neoplasms inhibits autophagy, as does oncogenic activation of PI3K/AKT and BCL2[36]. Likewise, monoallelic deletion of the autophagy regulator, beclin-1, is observed in 40%-75% of breast and ovarian human tumors[37], suggesting that autophagy functions to suppress tumor initiation. Accordingly, genetic inactivation of beclin-1 in mice predisposes their development of a variety of tumors, findings consistent with the notion that autophagy regulates cellular homeostasis and prevents tumor initiation[37-39]. Furthermore, activation of the transcription factor NRF2 elicits deregulation of autophagy due in part to aberrant accumulation of p62/SQSTM1 that can promote tumor formation[40-42]. Indeed, under tonic conditions, NRF2 interacts with Keap1, which targets NRF2 for ubiquitin-mediated degradation. The interaction between Keap1 and NRF2 can be prevented by the accumulation of p62/SQSTM1, thereby: (1) inhibiting the activation of autophagy; (2)stabilizing NRF2 expression, leading to its transcriptional activation[40-43]. Finally, emerging evidence suggests a role for autophagy in maintaining genomic integrity, as metabolic stress induced by loss of autophagy can promote DNA damage and chromosomal instability[44]. Indeed, when confronted with DNA damage, autophagydeficient cells exhibit diminished homologous recombination (HR) repair of damaged DNA that arises due to proteasomal degradation of checkpoint kinase 1 (Chk1)[45,46]. While non-homologous end joining (NHEJ) appears to be largely unaffected by autophagy inhibition, the diminished HR proficiency in these cells can render them more sensitive to DNA damage, especially if NHEJ is subsequently impaired[45,46]. Collectively, these findings identify important mechanisms whereby autophagy functions to suppress malignant transformation and tumor development.
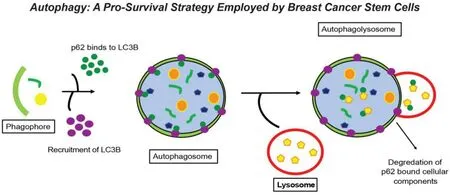
Figure 2. Autophagy: a pro-survival strategy employed by breast cancer stem cells. One of the pro-survival strategies employed by breast cancer stem cells during their acquisition of dormant states is autophagy, which facilitates the recycling of damaged or unnecessary organelles and/or proteins as a means to provide energy during periods of metabolic stress. Upon initiation of autophagy, the phagophore encircles those cellular contents targeted for autophagic degradation. LC3 is recruited to the phagophore and subsequently binds to the cargo adaptor protein, p62/SQSTM1. Upon doing so, a double membrane structure called the autophagosome forms and encircles cellular candidates for autophagic degradation. Subsequently, the autophagosome binds to the highly acidic lysosome to form the autophagolysosome, wherein p62/SQSTM1 bound cellular contents are degraded
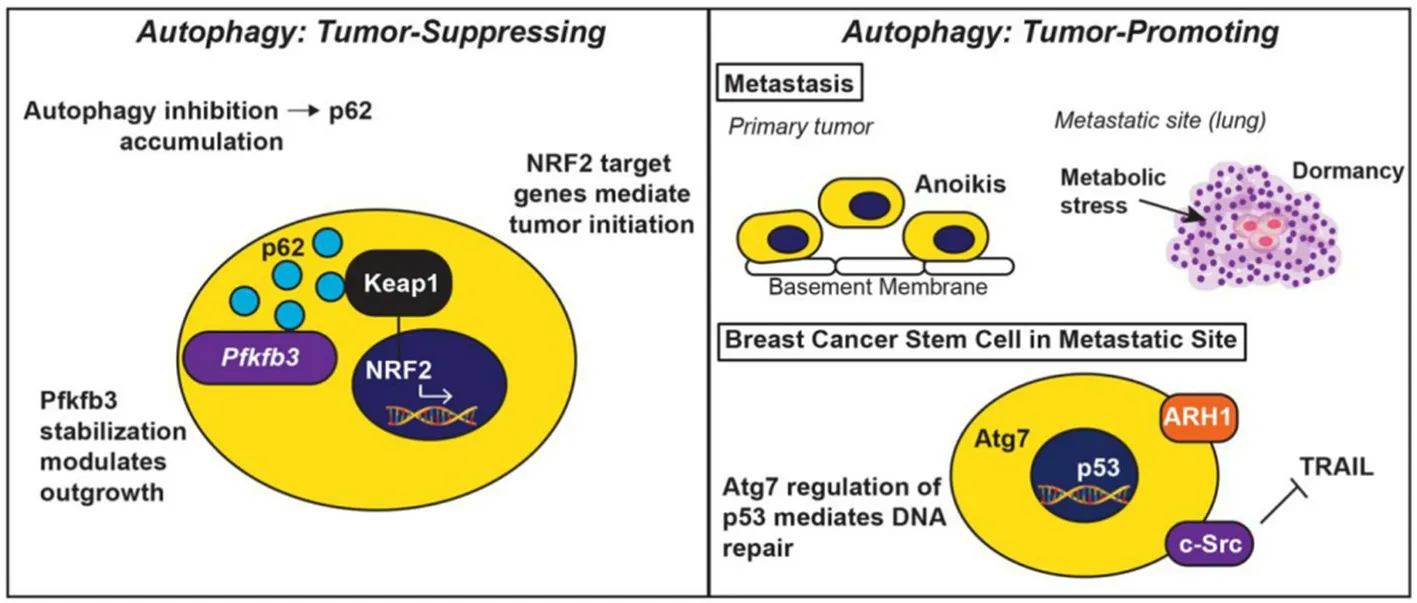
Figure 3. The tumor-suppressing and tumor-promoting activities elicited by autophagy. Autophagy functions to suppress tumor initiation (left panel), as well as to promote tumor development and progression (right panel). In early stages of tumor formation or during periods of metastatic dormancy, autophagy is tumor suppressive. Upon autophagy inhibition, p62/SQSTM1 accumulates and stabilizes Pfkfb3, leading dormant breast cancer stem cells to initiate metastatic relapse. Additionally, p62/SQSTM1 also inhibits the interaction between Keap1 and NRF2, thereby preventing NRF2-mediated expression of genes operant in tumor initiation (left panel). In stark contrast, autophagy provides established tumors with pro-survival phenotypes, including protection from anoikis and intrinsic cellular stressors encountered during metastatic dormancy. Likewise, autophagy protects breast cancer stem cells by ensuring for their resistance to the apoptotic stimuli housed within the metastatic microenvironment (e.g., Src-mediated TRAIL resistance), and to chemotherapeutic insults (e.g., Atg7-mediated p53 regulation of DNA repair). Finally, dormant cells can upregulate ARH1 to induce autophagy and promote the activation of pro-survival signaling systems that ensure for their survival
AUTOPHAGY AND TUMOR PROMOTION
In contrast to its tumor suppressing functions, autophagy can also serve as a tumor promoting process, particularly by: (1) enhancing the ability of DTCs to traverse the metastatic cascade; (2)inhibiting immunosurveillance by tumor infiltrating immune cells. During metastasis, cells shed from the primary tumor must invade through the extracellular matrix, intravasate into blood vessels, survive the turbid flow of the vasculature, extravasate, and finally colonize a distant metastatic site[47]. Not surprisingly, cells traversing the metastatic cascade experience a variety of cellular stressors and vastly different tissue microenvironments, including changes in the (1)composition of the extracellular matrix composition; (2)availability of nutrients due to alterations in vascular and lymphatic networks; (3)biomechanical properties of metastatic sites; (4)tumor immunosurveillance programs[48-50]. Importantly, autophagy activation protects DTCs during periods of metabolic stress encountered by anoikis and entry into foreign microenvironments[51-53], and by bouts of dormancy at distant metastatic sites[54,55].
AUTOPHAGY INHIBITS TUMOR IMMUNOSURVEILLANCE
Tumor immunosurveillance is a critical physiological process that inhibits the development and progression of mammary tumors. Accumulating data in the literature indicate that tumor immunosurveillance programs are an all-encompassing system that involves not only the adaptive immune system and cytotoxic effector pathways, but also the release of a complex set of cytokines and chemokines that coalesce to prevent tumor development[50]. Although the molecular mechanisms used by tumor cells to escape immunosurveillance are varied, recent evidence has implicated a role for autophagy in mediating this phenomenon. Indeed, autophagy activation has been shown to inhibit immune cell killing of tumor cells as a means to promote escape from immunosurveillance and DTC outgrowth[50]. In doing so, autophagy activation can target the activation of Signal Transducer and Activator of Transcription 3 (STAT3), a gene commonly dysregulated in breast cancer that also plays a prominent role in regulating the immune system[56]. For instance, the activation of autophagy can induce the phosphorylation and stimulation of STAT3 in tumors, an event that initiates cellular cross-talk between tumor and immune cells that ultimately suppresses Cytotoxic T Lymphocyte (CTL)-mediated lysis of tumor cells[56,57]. Likewise, autophagy can inhibit natural killer cell (NK)-mediated tumor cell killing by degrading granzyme B, a serum protease that is released by NK cells during NK-mediated cell killing[58,59]. Finally, breast cancer development and metastatic progression is critically dependent upon Epithelial-Mesenchymal Transition (EMT) programs[60]. Interestingly, autophagy is activated as carcinoma cells traverse the EMT program, with the resulting post-EMT mesenchymallike cells exhibiting elevated levels of autophagy relative to their pre-EMT epithelial-like counterparts. Importantly, EMT-mediated activation of autophagy inhibits CTL-mediated antitumor immunosurveillance in a beclin-1-dependent manner[50,61-63]. Collectively, these studies highlight the tumor intrinsic and extrinsic functions of autophagy, with the latter function, fulfilling an essential role in governing the fidelity of tumor immunosurveillance mechanisms.
DORMANT BCSCS AND AUTOPHAGY ACTIVATION
BCSCs can lie dormant for decades before recurring as metastatic lesions in breast cancer patients. During this time, disseminated BCSCs must survive nonpermissive tumor environments, while simultaneously maintaining their viability and the capacity for tumor initiation[5,64]. Emerging evidence implicates autophagy as an essential feature in maintaining the phenotypes associated with BCSCs, particularly their resistance to chemotherapies and hypoxic microenvironments[16,18,54,55,65-67]. The mechanisms whereby autophagy promotes BCSC survival at metastatic sites are varied and include the ability to confer resistance to apoptotic stimuli (e.g., Src-mediated TRAIL resistance in bone metastases[68]), to chemotherapeutic insults (e.g., DNA repair via Atg7 and p53 by Atg7[69]), and to cellular stressors[70]. Similarly, aberrant expression of ARHI (aplasia Ras homolog member 1) can elicit autophagy activation and modulate the survival of dormant cells in preclinical models of ovarian cancer, further implicating autophagy as an essential mediator of dormant cell survival[71]. Finally, tumor cells that possess defects in autophagy readily accumulate p62/SQSTM1, an event that alters p62/SQSTM1 function and contributes to tumorigenesis[17]. The aforementioned studies highlight the oncogenic and pro-survival activities of autophagy that contribute to tumor progression, as well as the acquisition and eventual emergence from metastatic dormancy. In the succeeding sections, we discuss the implications of targeting the dichotomous roles of autophagy in clinical settings.
CLINICAL TARGETING AND MODULATION OF AUTOPHAGY
Numerous clinical trials have aimed with varying degrees of success to inhibit or stimulate autophagy as a potential cancer therapeutic[35]. Despite significant investments in preclinical and clinical investigations, no FDA-approved drugs designed to modulate autophagy have been approved for the treatment of primary or metastatic breast cancers. This clinical deficit reflects the challenges associated with the dichotomous roles played by autophagy during mammary tumor development and metastatic progression, and with the inability of science and medicine to fully appreciate the downstream consequences of autophagy modulation in metastatic disease settings.
INHIBITORS OF AUTOPHAGY
At present, nearly 32 human clinical trials have been undertaken to assess the efficacy of autophagy modulating agents [Table 1], either administered alone or in combination with standard-of-care chemotherapeutics (www.clinicaltrials.gov). Pharmacological inhibition of autophagy in clinical settings is primarily accomplished using chloroquine, or a closely related molecule, hydroxychloroquine. Chloroquine functions to block autophagosome-lysosome fusion by preventing the acidification of the lysosome, thus inhibiting autophagy[72,73]. While the vast majority of studies include either chloroquine or hydroxychloroquine in combination with standard-of-care regimens, one recent study utilized a novel proteasome inhibitor, MLN9708, as a means to assess the impact of autophagy in conferring breast cancer resistance to the cytotoxic activities of doxorubicin[74]. Interestingly, administration of MLN9708 to breast cancer cells resulted in autophagy activation in a manner paralleling previous connections between proteasomal inhibitor and autophagy[75,76]. Moreover, MLN9708 enhanced the sensitivity of breast cancer cells to doxorubicin in a manner that was inversely correlated with the extent of autophagy activation[74]. As such, future studies need to assess the effectiveness of combining proteasomal and autophagy inhibitors with cytotoxic chemotherapies (e.g.,doxorubicin).
Additional translational insights into how autophagy inhibition impacts cancer cell survival has been accomplished using a combination of pharmacologic (e.g.,choloroquine and/or hydroxychloroquine) and genetic (e.g.,knockdown of autophagy associated genes) approaches. In general, these studies support the concept that inactivation of autophagy limits the development and spread of human cancers. Interestingly, recent evidence indicates that the molecular mechanisms underlying the cytotoxic activities of chloroquine and hydroxychloroquine are distinct from those employed to inhibit autophagy. Indeed, induction of lysosomal membrane permeabilization was insufficient to elicit apoptosis in cells treated with chloroquine. Rather, the cytotoxic activities of chloroquine were found to manifest subsequent to mitochondrial membrane permeabilization[77], and to reduced expression and activity of JAK3 and DNMT1[78]. Precisely how these alternative targets and activities attributed to chloroquine contribute to its clinical successes and failures remains an important line of research in the field of autophagy modulation.
STIMULATORS OF AUTOPHAGY
In light of the dichotomous activities autophagy plays during tumorigenesis, clinical investigation has also evaluated the impact of stimulating autophagy as a means to limit the growth and spread of cancers. Indeed, mTOR (mammalian target of rapamycin) is the primary pathway targeted pharmacologically as a means to induce autophagy in human breast cancers. For instance, several studies have investigated the importance of rapamycin[79], Everolimus[80,81], and Temsirolimus[80,82]as potential inducers of autophagy in clinical settings. In general, autophagy activation elicited in response to mTORC1 inactivation is a byproduct of the intended drug target, thereby producing synergistic cell killing in the form of autosis (i.e., autophagic cell death[83]). Along these lines, several clinical trials associated with mTOR modulation have sought to overcome endocrine resistance associated with hormone receptor positive breast cancer treatments. Unfortunately, single agent modification of autophagy by administration of mTOR pathway inhibitors has proven to be
highly ineffective at restoring endocrine sensitivity to estrogen receptor-positive breast cancers. Likewise, combining autophagy modulators with anti-estrogens has also failed to significantly improve the clinical course of these patients, with severe toxicities being associated with Everolimus[80]. Thus, similar to the strategy of autophagy inhibition, the clinical utility of autophagy activation to eliminate metastatic breast cancers awaits additional mechanistic and translational investigation.

Table 1. Cancer clinical trials evaluating autophagy modulation
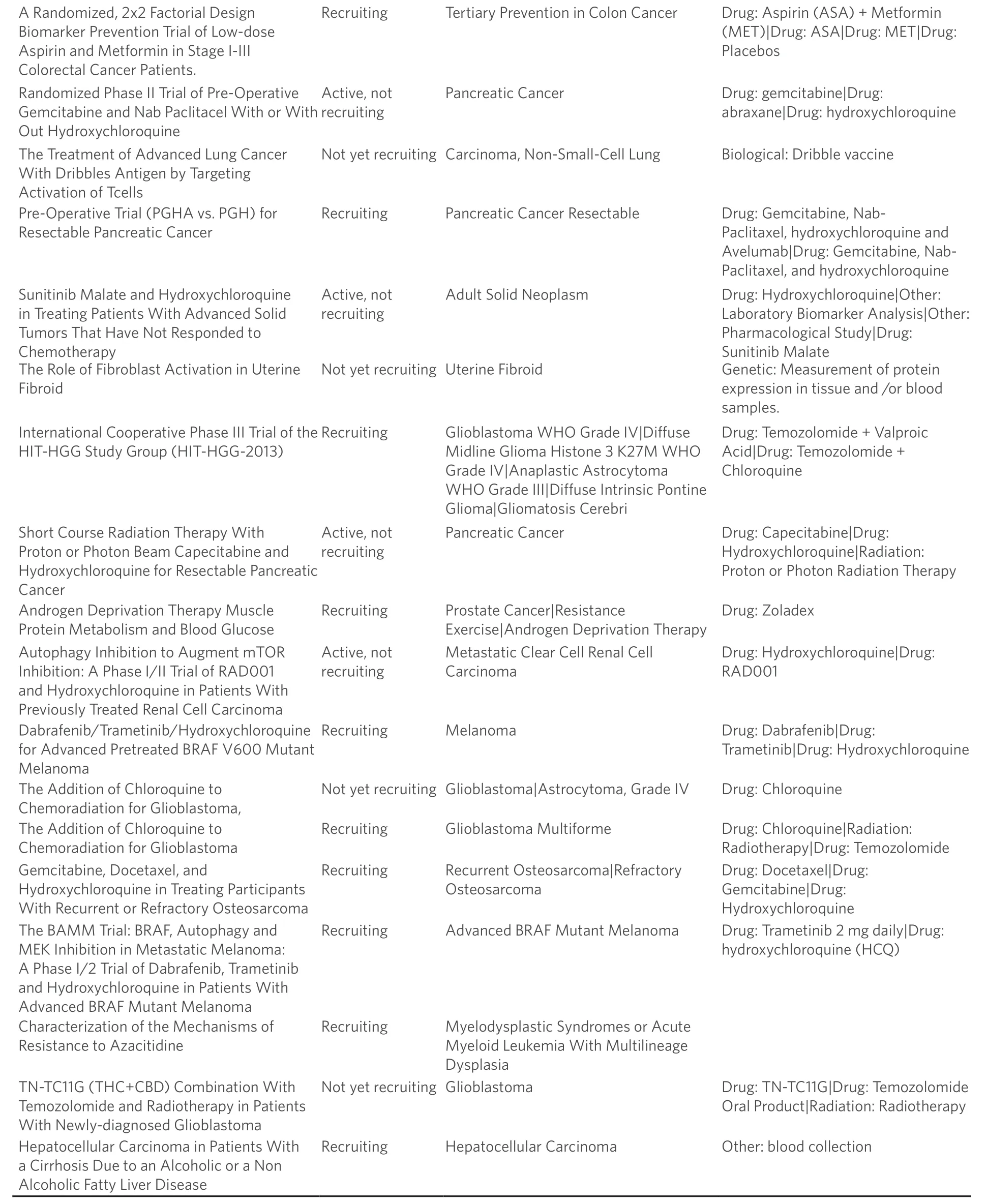
List of clinical trials (www.clinicaltrials.gov) that are currently active, recruiting, or not yet recruiting patients for clinical trials to study how autophagy modulation, primarily through chloroquine or hydroxychloroquine treatment, influences tumor growth and progression
CONCLUSION
Metastatic dormancy is mediated by BCSCs and responsible for the majority of breast cancer-associated deaths. An inherent property of BCSCs reflects their ability to activate a variety of pro-survival strategies to circumvent metabolic stress within the metastatic niche, and to overcome therapeutic insults mediated by chemotherapies and radiation. The activation of autophagy has proven to be a critical component of the pro-survival strategies employed by BCSCs, especially when confronted with nutrient deprivation, with inhospitable tissue microenvironments, with cytotoxic agents, and with dormancy-associated phenotypes. Indeed, preclinical evidence implicates important roles for autophagy modulation in the treatment of breast cancer. However, the paradoxical functions of autophagy to both suppress and promote tumorigenesis has clearly hampered the development and implementation of effective autophagy modulators for the treatment of metastatic breast cancer. Accordingly, several important avenues of basic and clinical investigation need to be achieved in order to generate effective autophagic agents. First, studies need to determine the extent to which chemotherapeutic drugs rely upon autophagy modulation when inducing their cytotoxic activities in target cells. Indeed, these so-called “off-target” effects on autophagy may underscore either directly or indirectly the extent to which a therapeutic regimen is effective, or alternatively, is rendered insensitive. Second, additional efforts need to be directed at identifying improved autophagy modulating drugs, particularly those that are effective against metastatic disease. Third, enhancing our understanding of how the tumor microenvironment impacts the targeting of autophagy-directed drugs is also warranted[84-86]. Finally, efforts directed at developing biomarkers capable of identifying patients most likely to benefit from autophagy modulation needs to be undertaken to minimize potential untoward side effects (e.g.,disease progression, emergence from dormancy, and metastatic relapse) of this course of treatment. Ultimately, addressing these research avenues will provide new inroads for strategies aimed at targeting autophagy vulnerability in BCSCs, and consequently, at eliminating metastatic relapse.
DECLARATIONS
Acknowledgments
Members of the Schiemann Laboratory are thanked for critical comments and reading of the manuscript.
Author’s contributions
Conception and study design: Flynn ALB, Schiemann WP
Drafted and revised the manuscript: Flynn ALB, Schiemann WP
Availability of data and materials
Not applicable.
Financial support and sponsorship
Research support was provided in part by the National Institutes of Health (CA236273) to Schiemann WP, and (T32GM008803 and T32CA059366) to Flynn ALB. Additional support was graciously provided by the METAvivor Foundation to Schiemann WP, and by pilot funding from the Case Comprehensive Cancer Center’s Research Innovation Fund, which is supported by the Case Council and Friends of the Case Comprehensive Cancer Center to Schiemann WP, and from the Case Clinical & Translational Science Collaborative to Schiemann WP.
Conflicts of interest
All authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Authors 2019.
 Journal of Cancer Metastasis and Treatment2019年5期
Journal of Cancer Metastasis and Treatment2019年5期
- Journal of Cancer Metastasis and Treatment的其它文章
- Operative treatment of metastatic breast cancer in the spine with regard to molecular phenotypes
- Chemotherapy-induced immunological breast cancer dormancy: a new function for old drugs?
- The lncRNA BORG: a novel inducer of TNBC metastasis, chemoresistance, and disease recurrence
- Stem cells, immortality, and the evolution of metastatic properties in breast cancer: telomere maintenance mechanisms and metastatic evolution
- Training and evaluation of a knowledge-based model for automated treatment planning of multiple brain metastases
- ESR1 alterations and metastasis in estrogen receptor positive breast cancer