
Monoclonal antibody pharmacogenomics in cancer treatment
2019-07-29 04:25ShigehiroYagishitaAkinobuHamada
Shigehiro Yagishita, Akinobu Hamada
Division of Molecular Pharmacology, National Cancer Center Research Institute, Chuo-ku, Tokyo 104-0045, Japan.
Abstract Conventionally, in the pharmacokinetic/pharmacodynamic analysis of small molecule compounds such as cytotoxic anticancer drugs, polymorphism analysis of genes related to absorption, distribution, metabolism,and excretion has been performed in addition to the analyses of blood concentrations of drugs.Such pharmacogenetic factors play an important role in predicting therapeutic effects and adverse events and in the proper use of drugs.With the recent launch of immune checkpoint inhibitors (ICIs) and the rapid development of antibody-drug conjugates (ADCs) currently underway, there is no doubt that antibody drugs, which are large molecule compounds, will become key drugs in anticancer drug treatment.However, the pharmacokinetic and pharmacodynamic analysis of antibody drugs is still not sufficient, and further elucidation of factors and mechanisms affecting their dynamics in the human body is necessary.Moreover, the pharmacogenomic factors of antibody drugs have not yet been fully studied.There are many factors that should be clarified, such as factors that regulate the host immune response in ICI therapy and the effects of ATP-binding cassette transporter and cytochrome P450 on the payload of ADCs.This review provides an outline of antibody drugs in cancer treatment and summarizes the pharmacogenomic factors of antibody drugs known to date.
Keywords: Pharmacogenomics, antibody drug, antibody-dependent cellular cytotoxicity, immune checkpoint inhibitor, antibody-drug conjugate
INTRODUCTION
An antibody is a humoral immunity factor produced by B cells as a biological defense against foreign antigens in the living body.Antibodies, i.e., immunoglobulins, are composed of a light chain and a heavy chain, and there are 2 types of light chains and five types of heavy chains[1].Immunoglobulins are classified into IgA, IgD, IgE, IgG, and IgM according to heavy chain subtypes, and in human organisms, IgG accounts for 80%.The IgG family includes IgG1, IgG2, IgG3, and IgG4 according to heavy chain subtypes.On the other hand, immunoglobulin receptors are expressed on neutrophils and macrophages and are composed of glycoproteins called Fc receptors.The Fc receptor has subtypes depending on the immunoglobulin to be bound, IgA binds to Fcα receptor, IgD binds to Fcδ receptor, IgE binds to Fcε receptor, and IgG binds to Fcγ receptor.The structure of the heavy chain of IgG differs in its binding affinity to Fcγ receptors, resulting in specific functional properties for each IgG antibody subtype.
For the immunoglobulin function, four major types of mode of action are known: (1) the neutralizing function by binding of antibody to the antigen; (2) the function of opening the cell membrane by activating complement [complement-derived cellular cytotoxicity (CDC)]; (3) the opsonizing effect taken up by phagocytes through binding of antibody to the antigen; and (4) antibody-dependent cellular cytotoxicity(ADCC) that occurs by binding the antibody to NK cells and releasing cytokines.All currently approved antibody drugs are of the IgG subtype and produce their antitumor effects through these functions.
The application of antibodies to cancer treatment was proposed by the bacteriologist Paul Ehrlich around 1900, but their clinical application became practical since the discovery of monoclonal antibody production technology by the hybridoma of Köhler and Milstein[2]in 1975.Monoclonal antibodies are characterized by having high antigen specificity, producing almost infinite antibodies by hybridomas, and carrying out the same analysis by producing the same antibodies.They also have advantages for anticancer treatment, such as a long half-life, high potency, and low off-target effect.
As a result, monoclonal antibody therapy has increased hope as a “magic bullet”.However, the initial development of monoclonal antibodies using mice or rats has been abandoned one after another because of their short half-lives and high immunogenicity.With such a background, several techniques for modifying antibody formulations have been developed such as chimeric antibody that genetically substitutes the highly antigenic constant region of mouse antibody, humanized antibody that substitutes human immunoglobulin except for the complementary-determining region (CDR) site, and fully humanized antibody.Currently, about 26 antibody drugs have been approved against cancer, and the monoclonal antibodies for cancer treatment will be reviewed from a pharmacogenomic perspective.
CANCER THERAPEUTIC ANTIBODY
As of August 2019, a total of 26 antibody drugs have been approved for cancer treatment by the US Food and Drug Administration (FDA) [Table 1].
The antitumor efficacy of antibody drugs is brought about by any of the 4 functions mentioned above.Of the 26 FDA-approved drugs, 20 are IgG1, one is IgG2, five are IgG4 isotype, and there are no drugs of IgG3 isotype.This is considered to be due to the fact that IgG1, 2, and 4 have a half-life of about 21 days,while IgG3 has a short 7-day half-life.The main features of each IgG subtype are shown in Table 2.ADCC activity, which is dependent on the avidity of IgG and FcγR, is strongest in IgG3, moderate in IgG1, and weak in IgG2 and IgG4[3].Therefore, a drug that exerts ADCC activity is composed of an IgG1 isotype and an IgG4 or IgG2 isotype for the purpose of neutralizing action or signal inhibition.
Many of the antibody drugs that have been marketed are made to have ADCC activity or ADCC activity and a neutralizing effect as their main antitumor effects.In the 2010s, development of antibody-drugconjugates (ADCs), in which cytotoxic anticancer drugs are bound to antibody drugs, and immune checkpoint inhibitors (ICIs) that cause binding inhibition of immune checkpoint molecules has been rapidly advancing.
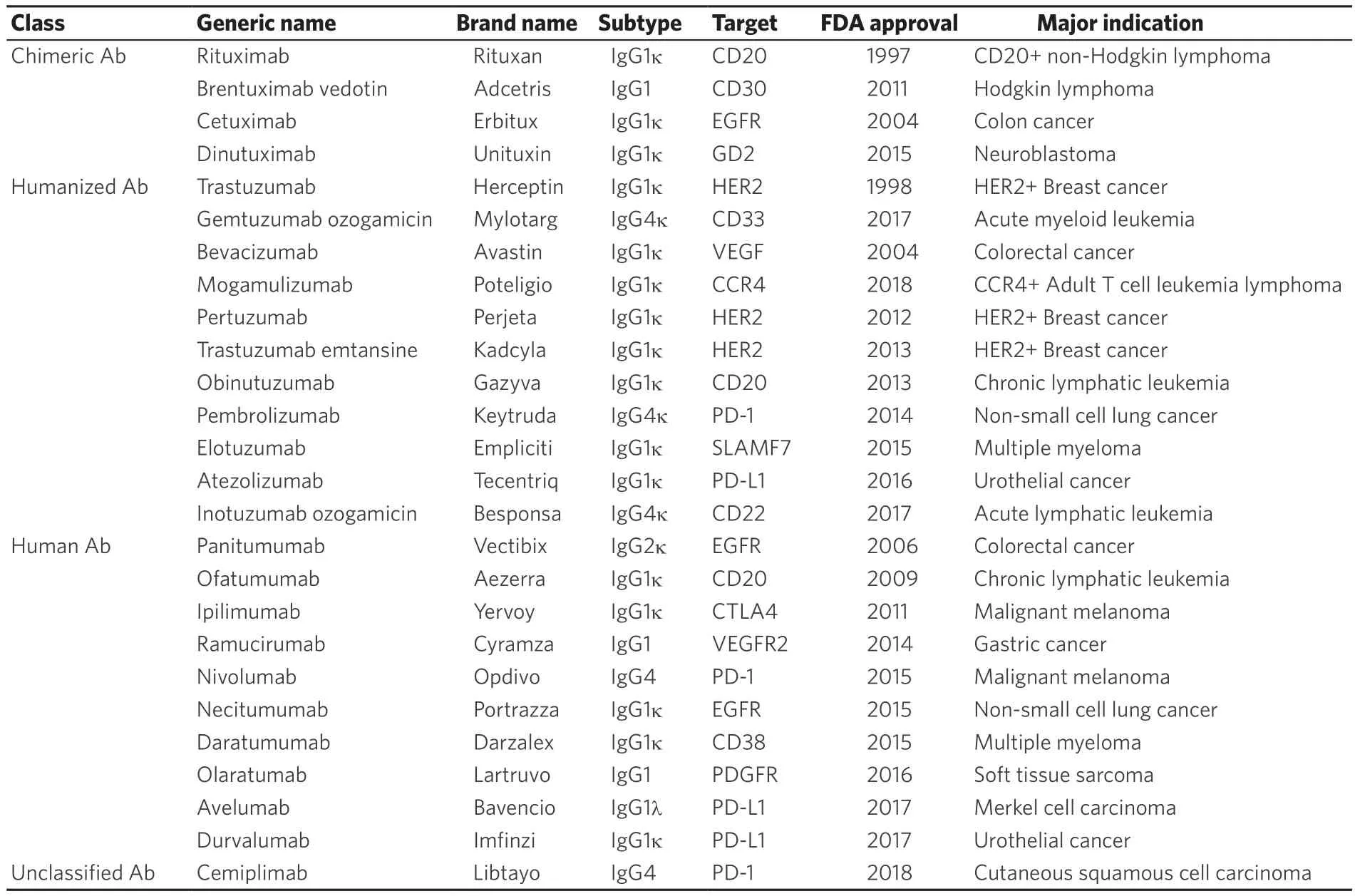
Table 1.Therapeutic antibodies for cancer treatment
PHARMACOGENOMICS OF CANCER THERAPEUTIC ANTIBODIES
Antibody drugs show anti-tumor effects by binding to antigensin vivo, but because they are proteins with a large molecular weight of about 150 kDa, they have complex pharmacokinetic and metabolic pathways that are completely different from small molecule compounds.Small molecule compounds generally have good membrane permeability and a large distribution volume (Vd) because they are distributed in cells[4].Since the effects of metabolism and excretion pathways are large for each drug, many pharmacogenetic studies have been conducted on the effects of polymorphisms of cytochrome P450 and ABC transporter on blood concentration levels of drugs.However, in the case of antibody drugs, the Vd is relatively small, they do not undergo metabolism such as by cytochrome P450, and the main elimination route is the digestion of amino acids in cells.For these reasons, they exhibit very different pharmacokinetics from small molecule compounds, and there are still many unknowns.A detailed description of the pharmacokinetics of antibody drugs was given by the critical review of Liming Liu, and this section outlines pharmacogenomic factors that affect the efficacy and pharmacology of antibody drugs[5].
Neonatal Fc receptor
The neonatal Fc receptor (FcRn) encoded byFCGRTwas assumed in the 1960s to be a receptor that protects IgG from catabolism by Roger Brambell[6,7].After cloning of FcRn by Simister & Mostov in 1989, analysis of knockout mice by Roopenian & Akilesh proved its function in 2007[8,9].The current understanding of FcRn is that blood circulating IgG is taken up by vascular endothelial cells and monocytes by pinocytosis and receptor-mediated endocytosis.Thereafter, IgG binds to FcRn in endosomes in an acidic environment (pH< 6.0), escapes lysosomal degradation, and is released again into the blood.If the blood IgG concentration is high, binding of FcRn to IgG is saturated, then lysosome-mediated IgG degradation is enhanced, and if the IgG concentration is low, IgG is bound to FcRn and recycled, thereby reducing IgG degradation[10].
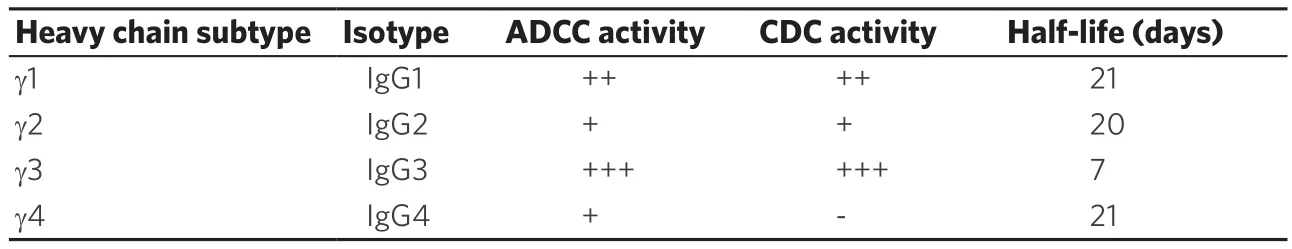
Table 2.Characteristics of IgG isotypes
Gene polymorphism of FcRn has been reported to be involved in FcRn expression, with VNTR of 37 bases occurring in the promoter region[11].The most common VNTR3/VNTR3 homozygous genotype is reported to have 1.66 times more FcRn expression than the VNTR3/VNTR2 heterozygous genotype[11].The levels of anti-TNF-alpha antibodies infliximab and adalimumab are lower in blood by 14% and 24%,respectively, among heterozygous cases compared to homozygous cases[12].On the other hand, as an antibody drug against cancer, in 94 cases that received cetuximab therapy, which is an EGFR antibody for colorectal cancer, a significant difference in distribution clearance and a tendency to a prolonged half-life were observed.There was no change in PK due to copy number variation (CNV) ofFCGRTgene[13].In addition, in 476 patients who received farletuzumab, an anti-folate receptor alpha antibody against ovarian cancer, there was no difference between PK and the area under the curve (AUC) of VNTR genotype and steady-state, and no change in PK due to CNV[14].As described above, there are few reports on blood concentrations of antibody drugs against cancer based on the FcRn genotype, and the relationship with efficacy is unknown.Further evidence is needed.
Antibody-dependent cellular cytotoxicity
An immunoglobulin including an antibody drug binds to an antigen at the Fab region, and the Fc region binds to Fcγ Receptor expressed on immune cells in the body.There are six types of FcγR: I, IIA, IIB,IIC, IIIA, and IIIB.Of them, I, IIA, IIC, IIIA, and IIIB are activated forms, and they mainly activate immune cells by phosphorylation of immunoreceptor tyrosine-based activation motif, which transmits activation signals into cells.On the other hand, IIB causes inhibitory functions via phosphorylation of an immunoreceptor tyrosine-based inhibitory motif[15].The ADCC activity as a mechanism of action of antibody drugs for cancer is considered to be the apoptosis of target cells by the release of perforin and granzyme from NK cells, mainly by the binding of the antibody drug to FcγRIIIA of NK cells.The binding strength of the antibody drug and FcγRIIIA is considered to be correlated with the ADCC activity.
Factors affecting the binding strength to FCγR include structural problems with the antibody drug and genetic polymorphisms affecting host immunity.Normally, the Fc region of immunoglobulins is glycosylated, and variations with or without terminal galactose, bisecting N-acetylglucosamine, sialic acid,and fucose at the root have been reported.For the ADCC activity, it has been reported that aglycosylation reduces ADCC activity, and addition of bisecting N-acetylglucosamine and removal of fucose increase ADCC activity.It has been reported that the addition of N-acetylglucosamine to mAb increases ADCC activity by 10-20 times[16].Moreover, fucose removal has undergone many technological developments,such as PotelligentTMtechnology (BioWa, Japan) and GlycomabTMtechnology (Glycart, Switzerland)based around cell lines engineered with altered glycosylation machinery, that increase ADCC activity by more than 100-fold[17,18].In the PotelligentTMcell line, both FUT8 gene alleles, which encode α1,6-fucosyltransferase, were disrupted by sequential homologous recombination.The GlycomabTMcell lines are stably transfected with the gene encoding 1,4-N-acetylglucosaminyltransferse III (GnIII), resulting in theexpressed antibodies bearing bisecting N-acetylglycosamine.Now, the PotelligentTMtechnology is used for the development of mogamulizumab, and the GlycomabTMtechnology is used for obinutuzumab.
DLBCL: diffuse large B-cell lymphoma; PFS: progression-free survival
On the other hand, there are gene polymorphisms ofFcGRas a host factor, and among them,polymorphisms ofFCGR2AandFCGR3Aare reported to be related to ADCC activity.A coding polymorphism in the extracellular domain ofFCGR2Ahas been described where a C>T substitution(rs1801274) changes the amino acid at position 131 from histidine to arginine (H131R).A second importantFcGRcoding polymorphism occurs in extracellular domain 2 ofFCGR3A; a T>G substitution changes valine to phenylalanine at position 158 (V158F, rs396991).
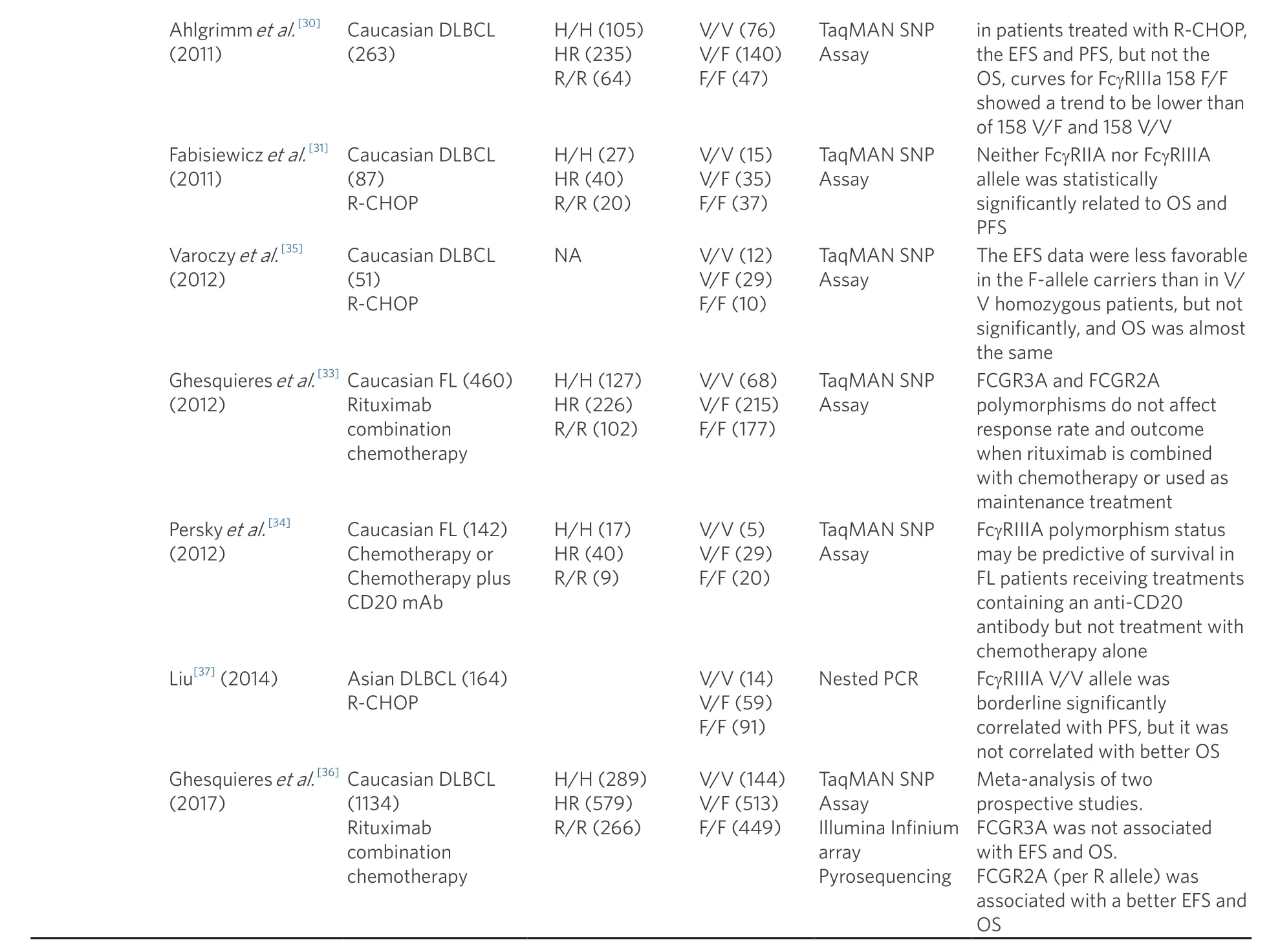
Although many studies have been conducted on trastuzumab and rituximab as to whether these gene polymorphisms affect the efficacy of antibody drugs, many conflicting reports have been published[Table 3].For trastuzumab, FcGR2A H/H and FcGR3A V/V are reported to be correlated with prolongation of progression-free survival (PFS) in metastatic breast cancer, FcGR2A H/H and FcGR3A V/V were reported to be correlated with pathological complete remission in the neo-adjuvant setting, and no obvious correlation was found between FcGR and overall survival in the adjuvant setting; thus, there is still no unified view[19-22].Similarly, rituximab has been investigated for its effect on drug efficacy against follicular lymphoma and diffuse large B cell lymphoma (DLBCL), but there are no reports of statistically significant effects, except in the early 2000s[23-37].As described above,FcGRSNPs and the ADCC activity of antibody drugs are clearin vitro, but their relationship with clinical efficacy is not clear.The reasons for this include the validity of the SNP verification method, the possibility that the number of cases needed to verify the effect of SNPon ADCC activity has not been collected, and the possibility that there are individual differences in the number or function of host immune cells that exert ADCC activity.The search for further mechanisms and factors involved in ADCC is desirable.
Immune-checkpoint inhibitors
In recent years, ICIs that activate the exhausted immune cells by inhibiting immune checkpoint molecules typified by CTLA4 and PD-1/PD-L1 pathways with antibody drugs have been key drugs for the treatment of malignant melanoma and non-small cell lung cancer.To date, 1 anti-CTLA4 antibody (ipilimumab),2 anti-PD-1 antibodies (nivolumab and pembrolizumab), and 3 anti-PD-L1 antibodies (atezolizumab,durvalumab, and avelumab) have been marketed.Anti-PD-1 antibodies have IgG1 isoforms, and the other ICIs have IgG4 isoforms.All drugs have anti-tumor effects, mainly due to neutralization or signal inhibition, with dramatic therapeutic effects in many cancer types such as malignant melanoma, NSCLC,and merkel cell carcinoma[38].However, ICIs have unclear drug efficacy biomarkers, and they cause immune-related adverse events (irAEs) that are different from the adverse events of conventional anticancer drugs.IrAEs occur due to the activation of host immunity by ICIs, causing immune response disorders and hormonal abnormalities in organs such as the thyroid, lungs, and pancreas.Although myelotoxicity and gastrointestinal toxicity are less likely than with cytotoxic chemotherapy, irAEs can sometimes be life threatening.Moreover, various biomarker searches for PD-1 inhibitors, which are frequently used in clinical practice, have been investigated.Tumor PD-L1 expression, tumor mutation burden (TMB), T cell receptor(TCR) repertoire, human leukocyte antigen (HLA), microbiome, etc.are attracting attention[39].ICIs activate host immunity by blocking the co-stimulatory/inhibitory pathway with antibody drugs, but the major signaling pathway of antigen-presenting cells and T cells is the binding of major histocompatibility complex (MHC) and TCR.Antigen-presenting cells present peptides produced from tumors as antigens,which are recognized by TCRs.From the above, peptides presented as antigens are factors on the tumor side, and MHC and TCR are factors on the host side.
Tumor antigen peptides that are factors on the tumor side include cancer testis antigens such as the melanoma-associated antigens (MAGEAs) and New York esophageal squamous cell carcinoma 1 that are expressed in tumors regardless of gene mutations, and tumor antigen peptides generated by tumor gene mutations.The number of tumor non-synonymous mutations is called the TMB, and it is attracting attention as an index of tumor immunogenicity, especially in melanoma and NSCLC that typically have high mutation burdens due to the mutagenic effects of ultraviolet light and cigarette smoking,respectively[40].In 2014, Snyderet al.[41]performed 64 whole-exome sequencing of melanoma patients treated with CTLA-4 antibody and showed significantly higher TMB in patients with stable or responsive disease for more than 6 months, and TMB more than 100 was associated with better overall survival[41].This result was confirmed by examination of CTLA-4 antibody against other malignant melanoma cohorts,and it was shown that the higher the TMB of non-small cell lung cancer, the better the response rate and PFS of PD-1 antibody[42,43].Since then, many cancer types have been shown to have a linear response rate with TMB, and it has become clear that TMB can help predict certain therapeutic effects[44].However,some cancer types do not follow this linear response.For example, merkel cell carcinoma and renal cell carcinoma are more sensitive than expected to TMB, whereas misamatch-repair (MMR)-proficient colorectal cancer is less sensitive than expected.That is, although TMB has some degree of correlation with the therapeutic effect, other factors are also assumed to be related to the therapeutic effect of ICIs.In addition, technical issues still remain, such as the fact that TMB does not have a confirmed calculation method and cut-off criteria, and whether analysis samples are performed with tumor samples or cell-free DNA.
Like TMB, DNA MMR defects (MMRds) are considered biomarkers of ICIs by causing somatic mutations.Leet al.[45,46]showed that pembrolizumab had a response rate of 53% in 86 cases with 12 cancer types oftumors with MMRds exhibiting micro-satellite instability (MSI), showing a robust efficacy regardless of the type of cancer[45,46].From these results, nivolumab and pembrolizumab were first approved by the FDA in 2017 with the genotype MSI-positive regardless of cancer type.
The HLA genotype is important as a host element.HLA class I is rich in sequence diversity of peptide binding sites.The HLA-I allele is encoded by three genes on chromosome 6 (HLA A, HLA B, and HLA C),and each of these variants constitutes a slightly different peptide.Chowellet al.[47]examined the genotype of HLA-I in 1535 patients treated with ICIs.They found that patients with HLA-B44 supertype showed prolonged survival, and patients with HLA-B62 supertype or somatic loss of heterozygosity at HLA-I showed reduced survival[48].Chowellet al.[48]analyzed the expression of MHC class I and II in tumor tissues in malignant melanoma treated with ipilimumab, nivolumab, or their combination.They found that 78 of 181 cases (43%) showed disappearance of MHC class I, which was correlated with initial tolerance of ipilimumab.In addition, MHC class II expression was observed in 55 of 181 cases (30%), and a correlation with the therapeutic effect of nivolumab was demonstrated[48].
As described above, several pharmacogenomic factors on the tumor side and host side are attracting attention as ICI efficacy biomarkers.It is difficult to establish a single biomarker for ICIs because there are multiple factors, such as blocking efficiency of the signal pathway and immune environment, on the host side.In particular, the host immune system is considered to be a large factor, and identification of a pharmacogenomic factor that is an index of host immune responsiveness is required.
Antibody-drug conjugate
Currently, antibody-antibody drugs (ADCs), which are obtained by linking a cytotoxic anticancer drug(called “payload”) to the antibody with a linker, have rapidly developed as a new antibody treatment strategy against cancer.The ADC is an innovative drug design approach that increases the local concentration of payload only around the target due to the high target selectivity of antibody drugs (called“bystander effect”).Drugs such as KS1/4-methotrexate and BR96-doxorubicin were developed as the firstgeneration ADCs in the 1980s, but their efficacy was not satisfactory[49,50].This may be due to insufficient titer of the drug itself, wrong target antigen selection, poor internalization efficiency of the antibody,poor tumor accumulation, linker stability too high/low, and immune response to mouse antibodies.Gemtuzumab ozogamicin, an anti-CD33 ADC, received FDA approval for the first time as an ADC product in 2000[51].However, subsequent clinical trials found no clinical effect and increased fatal adverse events,and approval was withdrawn[52,53].
In order to overcome the challenges of first-generation ADCs, appropriate target search and appropriate payload development are underway, and brentuximab vedotin (anti-CD30 ADC), ado-trastuzumab emtansine (anti-HER2 ADC), and inotuzumab ozogamicin (anti-CD22 ADC) were approved by the FDA as second-generation ADCs[54].However, the second-generation ADCs also have problems such as the presence of a few unbound types due to the instability of the linker, early clearance by becoming a free drug, and causing off-target toxicities.The development of these first- and second-generation ADCs showed that selections of appropriate tumor cell targets, payloads, antibodies, linkers, and payload binding sites were important points in drug discovery.In other words, if the appropriate target and an antibody with high binding power cannot be selected, the accumulation of ADC in the tumor will be reduced; if effective payload selection cannot be performed, sufficient cell killing effects cannot be achieved; if the stability of the linker is low, it will dissociate in the body and cause an off-target effect; and if the stability of the linker is too high, an effective bystander effect cannot be attained.
Based on the above, many third-generation ADC products are currently being developed [Table 4].In particular, in June 2019, the FDA approved Genentech’s anti-CD79b ADC, polatuzumab vedotin (Polivy),for relapsed/refractory DLBCL.This drug showed the surprising result that complete remission was obtained in 40% (16/40) in the polatuzumab vedotin and bendamustine plus rituzimab therapy group in the Phase 1b/2 trial (GO29365 study)[55].Currently, clinical trials are ongoing with hematological tumors such as follicular lymphoma and in combination with other drugs[56].Moreover, Daiichi Sankyo’s novel anti-HER2 ADC, Ds8201a (Trastuzumab Deruxtecan), is under development.In breast cancer, multiple phase 3 studies for patients with HER2-positive cancer (DESTINY-Breast01: NCT03248492, DESTINY-Breast02:NCT03523585, DESTINY-Breast03: NCT03529110), a phase 3 study for HER2-low cancer (DESTINYBreast04: NCT03734029), a phase 2 study for HER2-positive gastric (DESTINY-Gastric01: NCT03329690),lung (NCT03505710), and colon cancers (NCT03384940), and combined use with ICIs (NCT04042701,NCT03523572) are in progress.A recently reported Phase 1 trial for HER2-positive advanced breast cancer showed a surprising response rate of 59.5% despite a previous heavy treatment history with trastuzumab or T-DM1[57].On the other hand, as an adverse event, pneumonitis was reported in 20 cases, and 2 fatal cases also occurred.Currently, the FDA has granted Fast Track and Breakthrough Therapy designations, and early approval is expected.
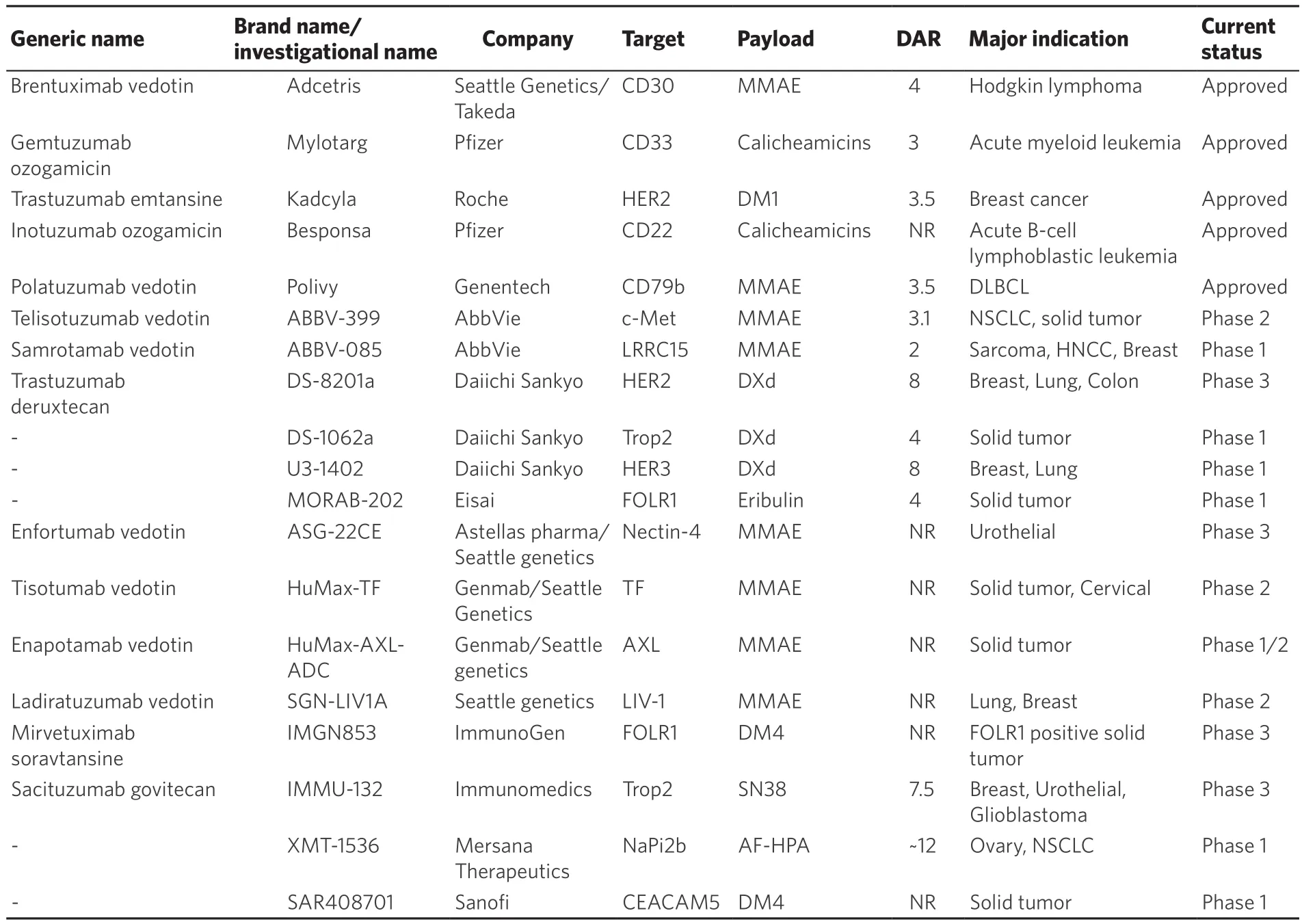
Table 4.Current status of antibody-drug conjugates
As shown in Table 4, there are currently 14 ADCs showing relatively good progress.Each drug targets not only HER2, but also HER3, Trop2, FOLR1, c-Met, AXL, and other molecules that are specifically expressed in tumors.Of 14 ADCs, 10 drugs used tubulin inhibitors (including 6 MMAE, 2 Maytansinoid DM4, 1 Eribulin, and 1 AF-HPA), and 4 drugs used a DNA topoisomerase I inhibitor (3 DXd, 1 SN38).In previous generations of ADCs, tubulin inhibitors were used in about 60% or more of payloads, but ADCs using a topoisomerase I inhibitor are increasing.In addition to improving the payload, each company is improvingthe drug-antibody ratio, the stability of the linker, and the cancer cell specificity, which have been issues with ADCs.In addition to the development of ADCs alone, their combination with ICIs is being actively developed.
Although it is expected that there will be a wider range of options for antibody therapy, the pharmacogenomic factors in ADCs are not clear so far.Since the antibody part is IgG1 in many ADCs, the above-mentioned FcγR polymorphism may have an effect.There is also concern about the effects of metabolic and excretory enzymes in the payload.For example, there are reports that ABCC1 overexpression is involved in drug resistance to trastuzumab emtanisine, and that ABCB1 is associated with the efficacy of gemtuzumab ozogamicin[58,59].In addition, MMAE, frequently used as a payload in many ADCs, is a CYP3A4 substrate, and eribulin is also a CYP3A4 substrate and an ABCB1 substrate.In other words, classical pharmacogenomic factors such as ABC transporter and CYP may affect metabolism and excretion of payloads and thus drug efficacy even in ADCs.
Moreover, recent ADC adverse events in clinical trials raise new concerns about immunogenicity.It has been reported that Ds8201a has a high incidence of pneumonitis, and this was the same case with Morab-202, which showed pneumonitis in 3 of 19 cases (15.8%)[60].Since the frequency of pneumonitis is not high with antibody drugs alone, pneumonitis may be a characteristic adverse event of ADCs.It is desirable to examine the pharmacogenomic factors of ADCs that are expected to be used more frequently in the future, including the enhancement of immunogenicity due to the linkage between linker and payload, and the possibility of affecting the responsiveness of host immunity.
As described above, ADCs have developed rapidly in recent years, but pharmacogenomic factors have not been fully studied.In addition to analyzing classical ABC transporters and CYP polymorphisms,pharmacogenomic analyses including host factors for characteristic adverse events such as lung injury are greatly needed.
FUTURE DIRECTION OF RESEARCH
There are still many black boxes in the pharmacokinetics of antibody drugs.Despite confirming doses several tens of times for the target occupancy in preclinical research, antibody blood concentrations in clinical practice may vary widely.Until now, it has been said that the pharmacokinetics of antibody drugs are not related to renal function, liver function or metabolic pathway, however, increased catabolism associated with organ dysfunction may affect the pharmacokinetics of antibody drugs.Furthermore,it is not clear whether the blood concentration of antibody drugs is correlated with the intratumoral concentration.It is necessary to identify the detailed pharmacokinetics of antibody drugs and the factors that affect the pharmacokinetics.
Another issue that must be considered is biosimilars.Unlike generic drugs, biosimilars cannot prove the identity of active ingredients.Therefore, at the time of approval, structural similarity is shown in a comparative quality study, PD and toxicity are shown in a comparative preclinical study, and PK, safety and efficacy are confirmed in a comparative clinical study.However, comparative clinical trials are only specifically designed to rule out clinically relevant differences in safety or efficacy between the biosimilar and the reference medicine, and to confirm biosimilarity.Differences in sugar chain modification and activity between lots have also been pointed out in the previous products, and it is still unclear whether biosimilars can exhibit sufficient pharmacokinetics and antitumor effects in clinical practice[61].In addition,it will be necessary to verify immunogenicity and pharmacogenomic differences.
CONCLUSION
The pharmacogenomics of antibody drugs is a complex area with more relevant factors than small molecule compounds.It is expected that pharmacogenomic factors related to drug efficacy and adverse events will be identified by comprehensively analyzing not only PK and PD, but also multiple factors such as host immune environment and genetic factors.
DECLARATIONS
Authors’ contributions
Wrote the manuscript and prepared the figures and tables: Yagishita S, Hamada A
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
 Journal of Cancer Metastasis and Treatment2019年11期
Journal of Cancer Metastasis and Treatment2019年11期
- Journal of Cancer Metastasis and Treatment的其它文章
- High-risk HPVs, microbiota and epithelial carcinogenesis: state of the art and research contribution of in vitro 3D models
- Novel immunotherapeutic approaches in head and neck cancer
- CXCR4 signalling, metastasis and immunotherapy:zebrafish xenograft modeI as transIationaI tooI for anti-cancer discovery
- Loss of the Krüppel-like factor 4 tumor suppressor is associated with epithelial-mesenchymal transition in colorectal cancer
- Histone chaperone FACT and curaxins: effects on genome structure and function
- AUTHOR INSTRUCTIONS