
1例斑驳病大家系临床表型及基因突变研究
2019-07-16 09:46:24葛宏松
安徽医科大学学报 2019年6期
葛宏松,李 越,张 成,漏 琼,李 明
斑驳病的最具特征的临床表现是额部、发际部呈三角形或菱形白斑,常伴有横跨发际的白色额发;此外白斑常见于上胸、腹部及四肢,偶见于面部、手足、背部等部位。白斑出生后即发生,终生稳定存在,部分患者的白斑周边、甚至中央可以出现不同程度和大小的色素沉着斑[1]。组织病理学显示,白斑部位黑素细胞完全缺失。临床上表现各异,尽管是同一个家系成员,其临床表现也不完全相同。斑驳病是一种罕见的常染色体显性遗传性皮肤病,由于黑素细胞先天性发育不良导致的色素脱失性疾病[2]。位于人类染色体4q12上的编码肥大/干细胞因子(stem cell actor, SCF)细胞表面跨膜受体KIT的基因突变(也称c-kit或CDll7基因),导致胚胎发育期黑素母细胞的增殖、分化和迁移发生障碍,从而引起斑驳病的发生[3]。到目前为止在斑驳病中,已有70多个KIT基因突变被报道[2-7]。该研究报道1例斑驳病大家系中存在新的KIT基因突变位点及外显不全表现。
1 材料与方法
1.1 家系情况1家3代14位成员,其中患者4例,见家系图1。先证者(Ⅲ:2),男,2月,父母非近亲结婚。查体:一般情况良好,发育正常。皮肤科专科检查:先证者出生时即发现前额可见菱形分布的白斑横跨发际线,白斑处头发为白发,前胸、腹部、四肢可见弥漫性分布且相互融合的大片状脱色素斑,部分白斑周边可见数个类圆形钱币大小的淡褐色色素沉着斑,面积超过50%。此外,左小腹部可见深褐色咖啡斑。家系其他成员调查:先证者外公及大姨前额、双小腿有大面积界限清晰的白斑,部分白斑内可见少许散在的色素斑,先证者姐姐双小腿有大面积界限清晰的白斑,先证者母亲未见明显皮损;Wood灯下可见大片状界清的脱色素白斑,同时白斑区内可见弥漫分布、程度不均的类圆形色素沉着斑。见图2。
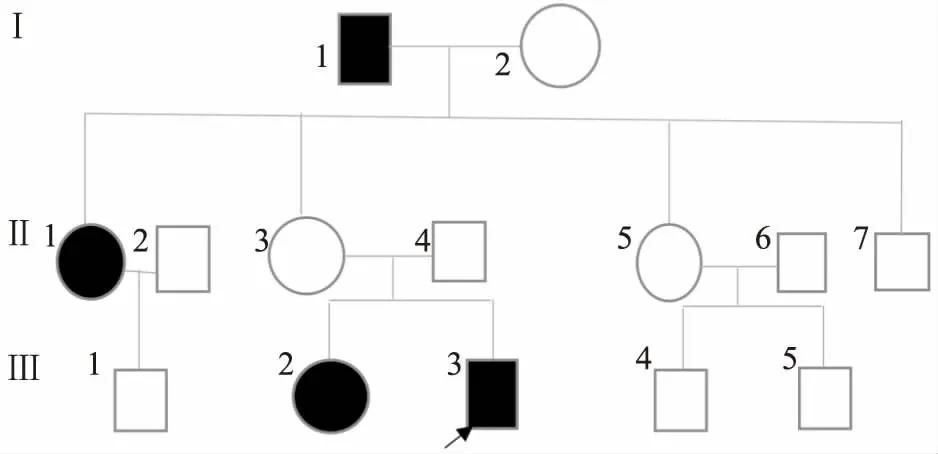
图1 家系图
1.2 外周血基因组DNA提取研究遵照赫尔辛基宣言,并由安徽省儿童医院伦理委员会审查通过。在征得所有患者以及正常人的知情同意后,采集所有家系成员及100例与本家系无关正常人的外周静脉血3 ml。用DNA提取试剂盒(购自美国普罗米加公司)提取基因组DNA。
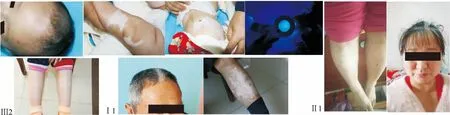
图2 典型病例的临床表现
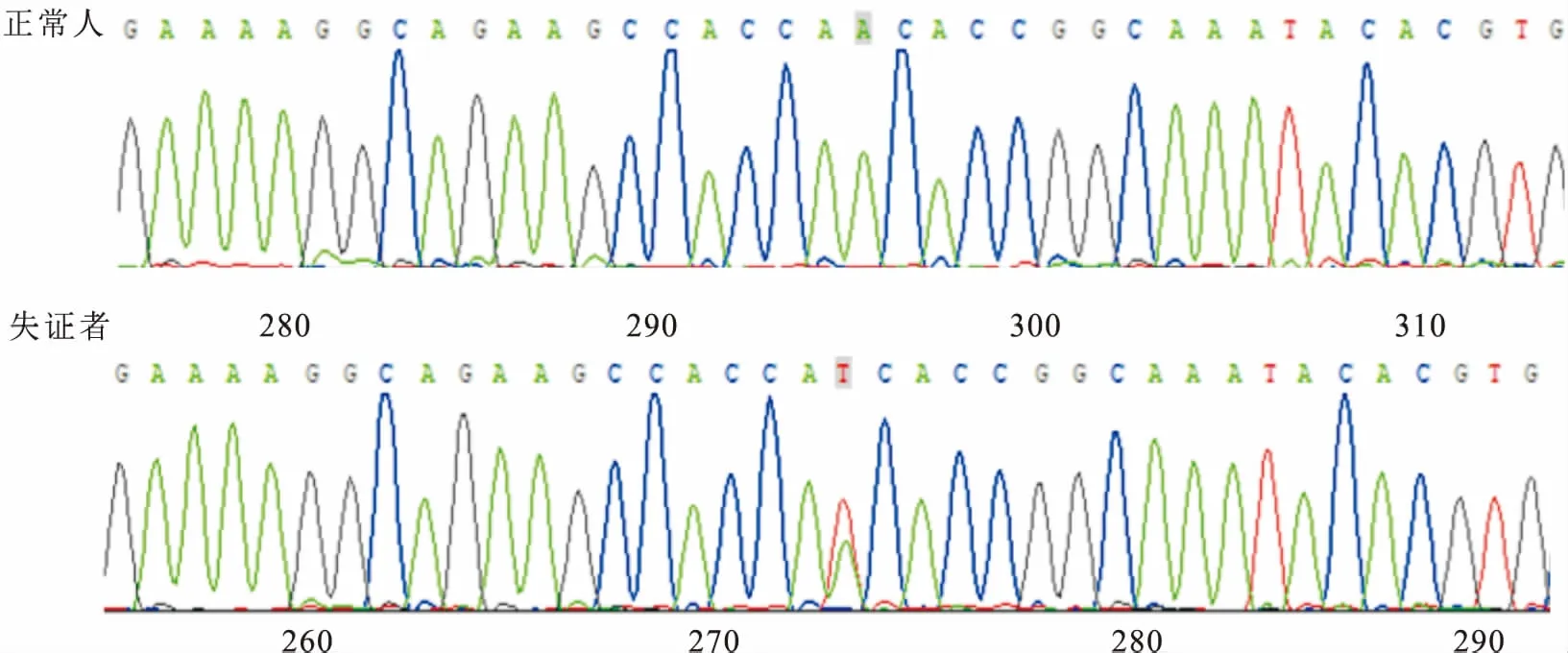
图3 KIT基因突变图
1.3 PCR扩增在线查取KIT基因mRNA序列经BLAST比对得到KIT基因的基因组序列(http://www.genome.UCSC.edu/),应用Primer3.0 (http://bioinfo.ut.ee/primer3-0.4.O/primer3/)在线平台设计针对KIT基因全部编码外显子的特异性引物进行PCR扩增,包括外显子及内含子交界50 bp的序列。采用25 μl的反应体系:10x缓冲液2.5 μl,10 mmol/L dNTP 0.5 μl,50 pmol/L双向引物混合物0.5 μl,TaqDNA聚合酶1.0 U,DNA模板1.0 μl (20 ng),加双蒸水至25 μl。PCR扩增程序如下:5 min升温至94 ℃,并在94 ℃变性持续30 s,于55~60 ℃退火,持续30 s,于72 ℃延伸,持续45 s,共38个循环,72 ℃终极延伸45 s。所有PCR产物经2%琼脂糖凝胶电泳检测验证。
1.4 DNA直接测序PCR产物送上海安百隆生物技术服务有限公司进行正向测序,测序结果与人类基因组KIT基因序列比较,如发现突变再行反向测序进行验证。
2 结果
成功扩增了KIT基因内所有外显子以及外显子与内含子交界区的基因序列。先证者的测序结果显示KIT基因外显子内第272位碱基A突变为T(c.272A>T);在家系内另3例患者及家系中正常成员先证者母亲也都检测到了该突变,而在家系中其他正常成员及100名正常对照经测序均未发现以上两个突变,因此排除了以上碱基改变为单核苷酸多态性的可能。见图3。
3 讨论
目前研究表明,斑驳病主要是由编码SCF受体的KIT基因突变引起,该突变导致受体功能的部分缺失[1]。人类KIT基因组位于4号染色体长臂区(4q12),DNA全长大约89 kb,包括21个外显子。SCF受体是一种受体酪氨酸激酶,斑驳病患者的KIT基因发生突变后,SCF受体功能下降,从而影响其介导的多条信号分子通路,进一步抑制胚胎期的成黑素细胞的增殖,导致胎儿出生后的色素形成异常,引起斑驳病。
目前被检测出KIT基因的突变类型有:点突变、不同位点发生碱基缺失或缺失、核苷酸剪接位点突变等,以上所有基因突变的结果均可导致KIT受体功能不同程度的改变,从而造成酪氨酸激酶功能的部分丧失或缺失,引起斑驳病特有临床症状的发生。迄今报道KIT基因突变约有70多种,大多数为无义或错义突变,其次是该基因的剪切位点突变。研究发现斑驳病临床表型的轻重与KIT基因突变的不同位点密切相关,KIT基因突变对SCF/KIT信号传导途径功能损害越大,其临床表型越严重。
近年来,国内外相继报道了一些KIT基因的错义突变,即c.1861G>A、c.1872G>A、c.860T>A、c.1861G>A和c.2017T>G等[2-7]。本例家系于第91位密码子出现错义突变c.272A>T(p.Asn91Ile),即属于此种突变,家系中患者之间显示出了较大的表型变异,其中最为严重的是先证者,先证者、家系内另3例患者及家系中正常成员先证者母亲也都检测到了KIT基因外显子内第272位碱基A突变为T(c.272A>T)存在该突变,此突变位点经文献检索未发现有类似报道。该家系中先证者母亲携带该突变,但目前为止未发现皮肤及毛发色素异常,可能为外显不全,原因可能在于除了KIT基因之外,可能存在其他调控基因影响疾病的发生,当然先证者的母亲随着随访的时间延长,也会出现轻症的临床表现。KIT基因该位点的突变致病分子机制值得我们进一步研究。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
当代水产(2021年9期)2021-12-02 01:34:54
基层中医药(2021年8期)2021-11-02 06:24:52
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
妈妈宝宝(2017年3期)2017-02-21 01:22:34
中国民族医药杂志(2016年1期)2016-05-09 08:34:41
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
