
减重手术后神经酰胺减少对脂肪重构的影响
2019-07-08 12:00汤海波朱利勇朱晒红
腹部外科 2019年3期
汤海波,朱利勇,朱晒红
(中南大学湘雅三医院胃肠二科 疝和减重代谢外科,湖南 长沙 410000)
脂肪组织中含有脂肪细胞、前脂肪细胞、成纤维细胞、血管内皮细胞和包括巨噬细胞在内的多种免疫细胞。脂肪组织对营养过剩或缺乏等改变做出快速、动态的反应[1],包括脂肪细胞的数量和(或)大小的动态变化,同时各种间质血管细胞发生数量和(或)功能的变化,有助于维持脂肪组织作为能量储存和内分泌器官的功能,这一生理过程被称为脂肪组织重构。脂肪组织重构是个复杂的过程,包含脂肪细胞的分化、增殖、衰老和死亡,并伴随着其分泌成分的改变和细胞外基质(extracellular matrix,ECM)的生成与降解等。与此同时,脂肪组织血管生成减少,免疫细胞如巨噬细胞的浸润[2]进一步导致全身低度慢性炎症和代谢紊乱,如胰岛素抵抗等。然而,在肥胖等病理生理条件的刺激下,脂肪组织分泌的细胞因子、激素和代谢产物的失调,打破了机体能量平衡稳态[3]。其中神经酰胺的代谢参与了如脂肪细胞凋亡和焦亡、分化和增殖、脂肪组织的慢性炎症和胰岛素抵抗,以及脂肪组织“褐变”等过程,在肥胖和胰岛素抵抗发生发展过程中发挥了重要作用。
一、神经酰胺的代谢与生理功能
神经酰胺的代谢(图1)主要包含了3种途径:①通过鞘磷脂酶水解细胞膜鞘磷脂而生成;②通过内质网从头合成即由丝氨酸棕榈酰转移酶介导的棕榈酰辅酶A和丝氨酸转化为3-酮基二氢鞘氨醇,然后进行一系列反应,最终产生神经酰胺;③补救合成途径:复杂的鞘磷脂也可以通过存在于酸性溶酶体中的各种酶降解生成神经酰胺。
神经酰胺由鞘氨醇和脂肪酸组成,根据其酰基链长度从C14∶0到C30∶0不等分为不同种类,由6种神经酰胺合成酶(CerS1~6)合成[4]。神经酰胺在不同组织的表达量及其种类均有所差别,其中和肥胖相关的特定CerSs及其对应的酰基链长度的改变很可能与代谢疾病密切相关。肥胖人群脂肪组织中CerS6 mRNA和C16∶0神经酰胺表达升高,且CerS6表达升高与胰岛素抵抗相关。相反,CerS6敲除(CerS6Δ/Δ)小鼠可以抵抗高脂饮食诱导的肥胖和糖耐量异常。此外,棕色脂肪组织中特异性敲除CerS6(CerS6ΔBAT)增加其对脂质的利用率[5](图2)。C16∶0神经酰胺可以直接或间接抑制脂肪酸氧化,电子传递链功能障碍产生活性氧,抑制肉毒碱棕榈酰基转移酶1(CPT1)活性,阻止脂肪酸进入线粒体氧化[6](图3)。

注:ER.内质网; SPT.丝氨酸棕榈酰转移酶;CerS.神经酰胺合成酶;SMase.鞘磷脂酶;SMS.鞘磷脂合成酶;CDase.神经酰胺酶;SphK.鞘氨醇激酶;SPPase.磷酸鞘氨醇磷酸酶图1 神经酰胺的代谢
二、神经酰胺介导脂肪细胞凋亡
已有多项研究表明神经酰胺能够激活诱导细胞凋亡的信号传导[7-9]。由神经酰胺介导的细胞凋亡机制可能和抗细胞凋亡蛋白B细胞淋巴瘤2(Bcl-2)的表达水平有关,Bcl-2定位于线粒体、内质网膜和核膜等细胞内氧自由基生成位点,被认为具有抗氧化和清除自由基的作用,由其编码的膜蛋白已证实在不同的试验条件下抑制细胞凋亡[10]。一项由Ortega等[11]进行的研究针对16例病态肥胖女性在接受腹腔镜下Roux-en-Y胃旁路手术前和术后2年,分别检测腹部皮下脂肪组织中全基因组mRNA的表达,其中术后Bcl-2相关蛋白A1(BCL2A1)的表达量不足术前的一半(P<0.001)。除此之外,脂肪细胞的凋亡可能与神经酰胺激活了转化生长因子激酶1(TAK1)的表达有关,选择性敲除TAK1的脂肪细胞中检测到细胞凋亡蛋白酶3(caspase 3)的含量明显升高。TAK1敲除的小鼠脂肪细胞数量减少,对高脂饮食或瘦素缺乏引起的肥胖具有抵抗力[12],这是由于TAK1作为核转录因子κB(NF-κB)的重要调节剂因而可以决定细胞的存亡[13]。最新的一项研究表明,TAK1也可以介导受体相互作用蛋白激酶(RIPK1)磷酸化进而促进细胞的程序性死亡,这一过程并不依赖于caspase和NF-κB途径[14]。综上,减重术后病人体内神经酰胺的水平下降,一方面通过降低Bcl-2的表达减弱了机体抗凋亡的水平,另一方面通过减少TAK1的激活而促进脂肪细胞的凋亡。当然,神经酰胺和脂肪细胞凋亡的关系可能并不仅仅依赖于此两种方式。
三、神经酰胺介导脂肪细胞焦亡
细胞焦亡,也称为细胞炎性坏死,和细胞凋亡同属于程序性细胞坏死,但细胞焦亡往往发生的速度更快,细胞凋亡蛋白酶通过切割一种叫做gasermin D(GSDMD)蛋白形成含有GSDMD氮端活性域的肽段,而后者会使细胞膜出现孔隙直至最后细胞膜破裂,同时会伴随着大量的以白细胞介素1β(IL-1β)为代表的炎性物质的释放,从而诱发炎性级联放大反应[15-16]。
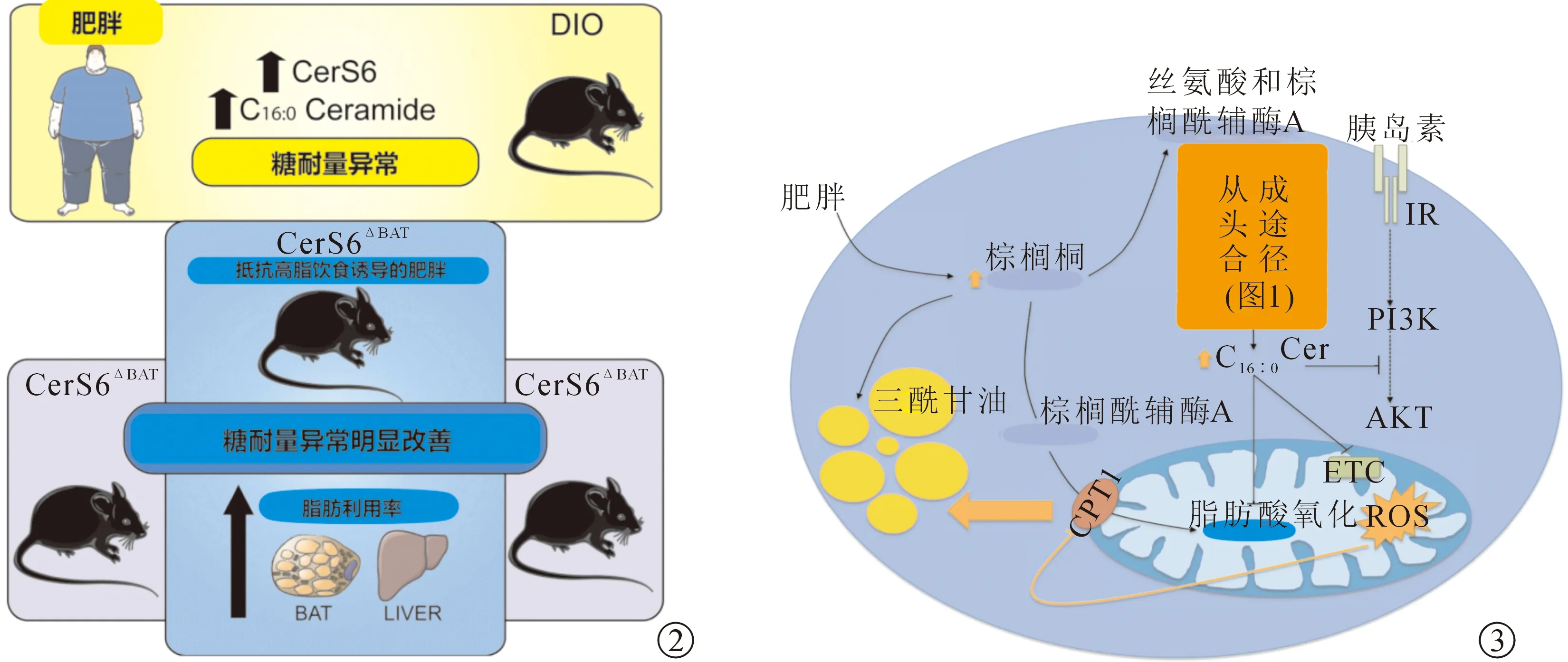
注:DIO.饮食诱导的肥胖小鼠;CerS6.神经酰胺合酶6;BAT.棕色脂肪组织;Cer.神经酰胺;PI3K.磷脂酰肌醇3激酶;AKT.蛋白激酶B;ETC.电子传递链;ROS.活性氧;CPT1.肉毒碱棕榈酰基转移酶1图2 C16∶0神经酰胺和肥胖 图3 C16∶0神经酰胺和脂肪酸氧化
众所周知,肥胖病人的脂肪组织存在程度不一的低级别慢性炎症,这源于其超过体重50%的脂肪含量以及超过100万个/g的免疫细胞,肥大的脂肪细胞同时增加了促炎脂肪因子的产生[17],这一过程和脂肪细胞的焦亡水平升高密切相关。Liu等[18]对小鼠脂肪细胞的研究发现,褪黑激素通过转录抑制GSDMD的表达,能缓解因脂多糖或肥胖诱导的NOD样受体蛋白3(NOD-like receptor protein 3,NLRP3)炎性小体激活,通过抑制NF-κB/GSDMD通路抑制小鼠脂肪细胞的焦亡。越来越多的证据表明神经酰胺在NLRP3炎性小体组装中发挥着重要作用,同时由于细胞焦亡释放的促炎细胞因子IL-1β又会反过来加强神经酰胺的作用[19],这一类似于“正反馈调节”的方式促进了脂肪组织的慢性炎症反应。多项研究表明减重手术降低了病人术后的血清、尿液中的神经酰胺水平并在术后相当一段时间内保持较低水平[20-23],这样一来,减重手术达到了和褪黑激素减少脂肪细胞焦亡类似的效果,在一定程度上缓解了脂肪组织的慢性炎症。
四、脂肪组织炎症和胰岛素抵抗
肥胖病人的脂肪组织慢性炎症除了与脂肪细胞焦亡释放促炎因子有关以外,还可能与脂肪组织中肿瘤坏死因子α(TNF-α)的变化有关[17],Jürets等[24]的研究包含31例病态肥胖的病人,分别检测其术前和术后1年的皮下脂肪基因表达情况,结果显示术后皮下脂肪中TNF-α相关基因的表达量高出术前2.9倍(P<0.001),而TNF-α在TAK1促炎症通路的上游发挥重要作用[14],术后神经酰胺的变化是否和TNF-α有关,目前仍无定论。
基质金属蛋白酶(matrix metalloproteinase,MMP)一般认为由结缔组织分泌并在细胞基质中发挥其作用,现在认为脂肪组织也会分泌这种蛋白酶,包括MMP-9和MMP-2[25-26]。ECM的重构是肥胖病理生理学的重要组成部分[26], MMP-9和MMP-2通过降解脂肪细胞间的基质蛋白,为脂肪细胞的肥大提供了有利条件。使用GEO数据库[27]在线分析来自于Arner等和Pelloux等关于减重手术前后皮下脂肪组织基因表达的研究,其公开的数据显示,接受了减重手术的病人其皮下脂肪组织的MMP-9相关基因表达量分别下调了5.58倍(P<0.001)和5.14倍(P<0.001)。Catalan等[28]的研究发现脂肪组织表达的MMP-9和无翅型MMTV整合位点家族成员5a(wingless MMTV integration family members 5a,WNT5A)呈正相关,和分泌型卷曲相关蛋白5(secreted frizzled-related protein 5,SFRP5)呈负相关。WNT5A通过非经典的WNT信号促进脂肪组织的炎症反应发生,同时也会介导中性粒细胞和巨噬细胞(M1占据优势)向脂肪组织的聚集[29-30](图4)。而在脂肪组织中过表达SFRP5,可以观察到循环中脂联素的浓度升高[28]。SFRP5作为关键的抗炎信号分子参与调控脂肪组织的慢性炎症,因其可以结合并拮抗WNT5A的作用从而抑制其下游非经典的促炎通路[30]。
WNT5A和SFRP5之间的平衡不仅可以调控脂肪组织炎症的发生发展,而且在胰岛素抵抗方面扮演重要角色[31-32]。神经酰胺通过介导脂肪细胞焦亡释放促炎细胞因子IL-1β,而MMP-9和IL-1β存在共表达关系[33],也就是说,神经酰胺可能通过此途径间接影响了WNT5A和SFRP5之间的平衡,除此之外,神经酰胺作为脂肪细胞重要的代谢中间产物,和胰岛素抵抗的关系同样值得探究。早在1998年的一项研究表明,C2-神经酰胺(一种人工合成的水溶性神经酰胺)可以激活丝/苏氨酸蛋白激酶(AKT)[34-35],而后者是胰岛素受体信号传导途径的关键蛋白激酶。就目前而言,在神经酰胺诱导的胰岛素抵抗机制方面,关于骨骼肌和肝脏的研究证据比较充足[36],而对于脂肪组织胰岛素抵抗的相关研究也正在加速进行。Bachnio-Zabielska等[37]报道了肥胖合并糖尿病病人的脂肪组织神经酰胺的含量与胰岛素抵抗指数(HOMA-IR)的相关性,与肥胖非糖尿病病人相比,肥胖合并糖尿病的病人脂肪组织神经酰胺的含量明显升高[38]。见图5。
五、脂肪组织“褐变”
脂肪组织一般有三种类型:白色、棕色和米色脂肪[40]。这三种类型的脂肪组织都与身体的其他器官包括皮肤、肝脏、胰腺、骨骼肌和大脑等广泛“交流”以维持能量平衡[41]。众所周知,白色脂肪负责存储脂类,棕色脂肪通过燃烧脂类、葡萄糖等释放能量。米色脂肪正是由白色脂肪在特定条件下转化生成,其细胞呈多房性外观(含有多个脂滴),并增加解耦蛋白1(uncoupling protein 1,UCP1)的表达[40]。在此过程中,这些通常储存能量的脂肪细胞变成了释放能量的脂肪细胞。Chaurasia等[38]研究认为,鞘脂类是脂肪组织产热的重要调节剂,其代谢中间产物神经酰胺促进了脂肪组织以三酰甘油等脂类的形式储存能量。Xia等[42]对过表达神经酰胺酶的转基因小鼠进行研究发现,脂肪细胞的形态发生了变化,同时改善了全身糖代谢以及脂质在肝脏的异常沉积。Jiang等[43]研究表明异位神经酰胺抑制脂肪组织的褐变,提示内源性神经酰胺可能是脂肪细胞功能的自主调节因子。Chen等[44]通过对小鼠施行减重手术,观察其术后棕色脂肪组织的变化,结果显示在术后4周,Roux-en-Y胃旁路术组棕色脂肪的体积和活性较术前以及其他手术方式组有明显增长(P<0.001)。
米色、棕色脂肪细胞燃烧脂质、释放能量涉及到细胞“动力工厂”——线粒体数量的增多和活性增强。线粒体中已经检测到可以参与神经酰胺生物合成和降解的酶[45],迄今报道的大多数研究也表明,细胞内神经酰胺含量的增加会干扰线粒体的能量代谢过程,从而损害线粒体功能。此外,神经酰胺诱导的线粒体呼吸异常与活性氧的产生和随后的氧化应激有关[46]。因此,减重手术降低了血清及脂肪组织中的神经酰胺浓度,在很大程度上减轻了神经酰胺对脂肪组织“褐变”的抑制作用,这样就有更多的棕色、米色脂肪细胞通过特异性表达UCP1使线粒体氧化磷酸化过程解偶联,从而促进游离脂肪酸氧化产生的能量以热量形式散发[47]。

图4 正常情况下,脂肪细胞数量增多、体积增大过程中同时伴随适当的血管生成和ECM重构,将细胞数量、体积限制在一定范围内(A);脂肪组织的病理性扩张时出现血管生成不足、ECM重构紊乱,从而导致随后的组织缺氧,M1巨噬细胞占据优势,导致与全身胰岛素抵抗密切相关的炎症表型(B) 图5 神经酰胺是针对多种肥胖信号(如饱和脂肪酸,satFA)、脂多糖(LPS)或促炎细胞因子(如肿瘤坏死因子,TNF),通过增加鞘磷脂生物合成或循环而产生的。神经酰胺可以激活蛋白磷酸酶2A(PP2A),从而去磷酸化和抑制丝/苏氨酸蛋白激酶(AKT),阻断胰岛素信号传导,导致胰岛素抵抗;另一方面通过激活蛋白激酶Cζ(PKCζ),也导致AKT通路的抑制;脂联素与其受体结合刺激神经酰胺酶(CDase),使神经酰胺降解为鞘氨醇,然后进一步磷酸化为鞘氨醇-1-磷酸(S1P)
六、结语
脂肪组织重构的机制涉及多种组织(如脂肪、骨骼肌、肝脏、中枢神经等)间的相互“交流”,多种细胞的病理生理改变,减重手术后机体神经酰胺的减少可能在该过程中扮演关键角色,这对于进一步理解手术“减重”、“降糖”的分子机制有重要意义,同时有助于开发新的治疗方式来预防和控制肥胖相关的代谢疾病。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
医学综述(2022年7期)2022-04-19
昆明医科大学学报(2022年1期)2022-02-28
医学综述(2021年24期)2022-01-11
中老年保健(2021年9期)2021-08-24
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2020年12期)2021-01-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国心血管杂志(2020年2期)2020-05-15
安徽医科大学学报(2020年1期)2020-02-14
