
生化药注射剂的质量评价策略与技术要点*
2019-07-08 10:48郑璐侠史芳亮吕晶张颖邵泓陈钢
医药导报 2019年7期
郑璐侠,史芳亮,吕晶,张颖,邵泓,陈钢
(上海市食品药品检验所,上海 201203)
生化药品是指从动物器官、组织、体液、分泌物中经前处理、提取、分离、纯化等制得的安全、有效、质量可控的药品,主要包括蛋白质、多肽、氨基酸及其衍生物、多糖、核苷酸及其衍生物、脂、酶及辅酶等。生化药注射剂是注射剂中较特殊的一类,按照化学药品质量控制方式很难控制其质量,与普通化学药品注射剂相比具有以下3个特殊性:①原材料大多来自生物体,来源复杂,组成不明确,本身具有不均一性,结构确证难;②生产涉及器官、组织、体液、分泌物的提取、分离和纯化等过程,仅通过终产品的质量标准很难有效控制质量;③生化药品需用效价测定、体外细胞活性实验、生物检定法等生物分析技术证实其生物活性,比理化测定具有更大的可变性。2017年度《国家药品不良反应监测年度报告》显示,按照给药途径统计,2017 年药品不良反应/事件报告中,静脉注射给药占61.0%,其他注射给药占3.7%,口服给药占32.0%,其他给药途径占3.3%。与2016 年相比,静脉注射给药占比上升1.3%,其中有多个生化药注射剂被提及,包括复方氨基酸(18AA) 、脂肪乳、丙氨酰谷氨酰胺和门冬氨酸钾镁等[1]。
近年来,随着药品监管体制改革的深入推进,国家有关部门陆续出台了一系列与生化药注射剂相关的新法规和政策。2008年1月8日,国家食品药品监督管理局颁布了《多组分生化药注射剂基本技术要求(试行)》,提出了多组分生化药注射剂审评中的重点关注点和相应的技术要求。2017年3月16日,国家食品药品监督管理总局(CFDA)药品审评中心发布了药品生产质量管理规范(good manufacturing practices,GMP)生化药品附录,作为《药品生产质量管理规范(2010年修订)》配套文件,旨在对生化药品的监管制定更具体、更严格的监管措施。2017年10月8日,中共中央办公厅国务院办公厅印发了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,明确要求严格注射剂药品审评审批和开展药品注射剂再评价。2017年12月22日,CFDA 药品审评中心组织起草了《已上市化学仿制药(注射剂)一致性评价技术要求(征求意见稿)》,明确了化学仿制药(注射剂)一致性评价的范围和技术要求。基于这样的时代背景,笔者针对生化药注射剂这一类特殊的高风险品种进行质量评价策略和研究要点讨论,以期为提升生化制药行业整体水平和药品质量提供借鉴和参考。
1 生化药注射剂现状
《中华人民共和国药典》2015年版共收载生化药注射剂67个,主要包括5大类:①氨基酸类,如复方氨基酸注射液等;②多肽、蛋白和酶类,如缩宫素注射液、注射用乌司他丁、注射用门冬酰胺酶等;③多糖类,如肝素钠注射液等;④核苷类,如注射用阿昔洛韦等;⑤多组分类,如垂体后叶注射液等。目前,国内生化药注射剂主要存在如下问题。
1.1同一品种批准文号过多 我国已批准的部分生化药注射剂品种的注册文号,存在同一品种有几十甚至上百个文号现象。如利巴韦林注射液,221家企业共有276个批准文号。根本原因在于主要以仿制为主,大多是低水平重复申报,长期存在同质化严重、产能过剩和恶性竞争现象,影响产品质量。还有一些生化药注射剂品种存在只占用文号,并不实际生产的现象,如注射用抗乙肝免疫核糖核酸,30家企业拥有40个批准文号,但调研后发现,近年内没有一家生产。
1.2立题合理性不足 生化药注射剂的立题合理性表现在更改剂型上。如在醋酸奥曲肽注射液的质量研究中发现,注射用醋酸奥曲肽与醋酸奥曲肽注射液相比,有关物质含量较高,稳定性较差,基本不含醋酸盐,生产工艺较繁杂,生产成本与风险也较高,且原研企业无此剂型,剂型的合理性需要进一步论证[2]。
1.3处方工艺与原研不一致 国内注射剂仿制药中成盐形式或辅料种类、用量与原研不符的现象较常见,在生化药注射剂中也有体现。如在注射用胸腺法新的质量研究中发现,原研药为胸腺法新的醋酸盐,但国内部分企业生产的样品中未检出醋酸盐,与原研药不一致,其成盐稳定性需要进一步考察论证。国内部分企业制剂处方中甘露醇用量明显低于原研制剂,非等渗溶液,易加剧注射部位不适;还易导致样品水分升高,增加质量隐患,应对处方合理性进行再确认。
1.4标识规格与实际生产不符 生化药品的标识规格多与常规化学药一致,以质量计,但其实际生物活性往往需采用效价单位表示方法(即U·mg-1),来反映生化药品单位质量中具有的生物活性高低。笔者在硫酸鱼精蛋白注射液的质量研究中发现,虽然该注射液标识规格为5 mL:50 mg或10 mL:100 mg,但国内制剂生产企业实际生产时按效价投料,并非按质量投料。具体来说,就是国产硫酸鱼精蛋白原料的效价水平可以达到140~150 U·mg-1,而制剂生产时是按照每毫克硫酸鱼精蛋白中和肝素100 U的效价进行投料折算,则规格为5 mL:50 mg的注射液实际投入硫酸鱼精蛋白33~36 mg,仅相当于标示量70%。随着企业生产工艺的提高,如果所生产或使用的原料效价提高,那么投入相同效价所需原料的质量就会相应地进一步减少。因此,该制剂并未达到规格所标识的主成分质量,与进口制剂标识的相同规格实际上存在差异。
1.5源头质量控制薄弱 《中华人民共和国药典》2015年版收载的常见生化药品既有来源于猪、牛、羊脏器及人尿液等,还有鱼成熟精子、牛血清等,这些原材料收集、处理、保存等基本不在药厂完成,不符合GMP管理要求,但其来源和质量优劣又是决定后续生化药品质量非常重要的关键点。如在鹿瓜多肽注射液的质量研究中,本实验室以核酸分子鉴定技术为基础,建立了鹿骨中梅花鹿及马鹿源性成分的鉴别方法[3],在部分企业的鹿骨原材料中除梅花鹿成分外同时检出马鹿成分,提示该企业鹿骨原材料有被马鹿源性成分混杂或污染的情况,其制剂存在潜在风险。
1.6物质基础不明,质量有待提高 生化药注射剂尤其是多组分生化药注射剂,由于来源复杂,提取物大多物质基础不明确,部分品种结构上具有异质性,尚无法达到批间完全一致性。生产企业往往以产品达到现行国家标准为主要目的,进行质量标准研究和提高的主动性较差,重视程度不高。这些原因均导致生化药注射剂质量标准很难真实反映产品间差异,整体质量标准水平有待完善。如笔者在注射用核糖核酸质量研究中发现,现行标准中缺少对核糖核酸片段长短、杂蛋白和苯酚残留的控制要求,从而会影响标准中紫外吸收系数法测定含量结果的准确性,有必要对质量标准进行提高[4]。
1.7包材质量存在隐患 药品包材是药品不可分割的组成部分,直接关系药品质量。玻璃是注射剂最常用的包装材料之一。基于成本等因素考虑,我国注射剂药用玻璃包装材料多数以钠钙玻璃和低硼硅玻璃为主,与国际上采用的中性玻璃的质量存在差距[5]。笔者对6家生产企业的注射用胸腺法新的包材玻璃中13种常见元素迁移和基于胶塞配方对其中的酚类有机化合物迁移进行考察,研究显示,有2家产品分别出现了抗氧剂二丁基羟基甲苯(butylated hydroxytoluene,BHT)和硫化剂对特辛基苯酚向药品迁移的现象,包材质量有待提高。
2 生化药注射剂的评价策略
美国食品药品管理局(FDA)对“一致性”的定义为:活性成分、剂型、规格、给药途径和适应证与参比制剂一致。FDA橙皮书共收载生化药注射剂22个品种(表1)。国内已上市的生化药注射剂既有FDA橙皮书上已收载的仿制药,也有一些根据中国国情开发的国外没有的特殊生化品种。因此,与化学药品注射剂的评价策略不尽相同,笔者认为,不应统称为一致性评价,而是应该根据品种的特点,进行一致性评价或再评价(图1)。
对于其中结构明确、物质均一、相对分子质量较小的化合物,如果有完整的安全和有效性数据或被FDA橙皮书收载,如右旋糖酐铁、更昔洛韦、盐酸精氨酸注射液等,应按照化学药品注射液要求进行一致性评价执行。否则应进行再评价,如肝水解肽注射液,虽然相对分子质量不大,但由于物质基础不明确,则应进行再评价。对于相对分子质量较大的化合物,如蛋白、多糖、酶等药物,如果结构明确,有完整的安全和有效性数据或被FDA橙皮书收载,如胰岛素、卵泡刺激素、肝素钠注射液等,可进行一致性评价,否则也应进行再评价。
对于结构不明确,物质基础不一致,又没有完整安全和有效性数据,同时也不在FDA橙皮书中的生化药注射剂,是否值得进行再评价,笔者认为关键在于临床是否有效。生产企业应该客观评价自身产品并进行荟萃分析,即在世界范围内收集对某一疾病疗效的小样本,以及单个临床试验的结果,对其进行系统评价和统计分析。如果有效,可以继续做药学再评价等,否则建议放弃。需要强调的是,对于那些结构不明确的生化药注射剂,只要临床有效就值得去坚持,如胰岛素注射剂,该品种的发现和临床应用已有近百年历史,但在临床刚开始使用时纯度并不高,人们对其结构亦不清楚,从1921年首次提取到胰岛素应用于临床,一直到20世纪80年代初才运用重组技术生产高纯度人胰岛素,中间经历了60多年时间,现在对其结构已经掌握到可以进行改构的程度,以根据实际需求达到快速起效或缓释的作用。这就是一个典型生化药走过的历程,从发现、提取、结构确认,到高纯度、最后可以按需改构,支撑它走完这一历程的重要因素就是临床疗效。
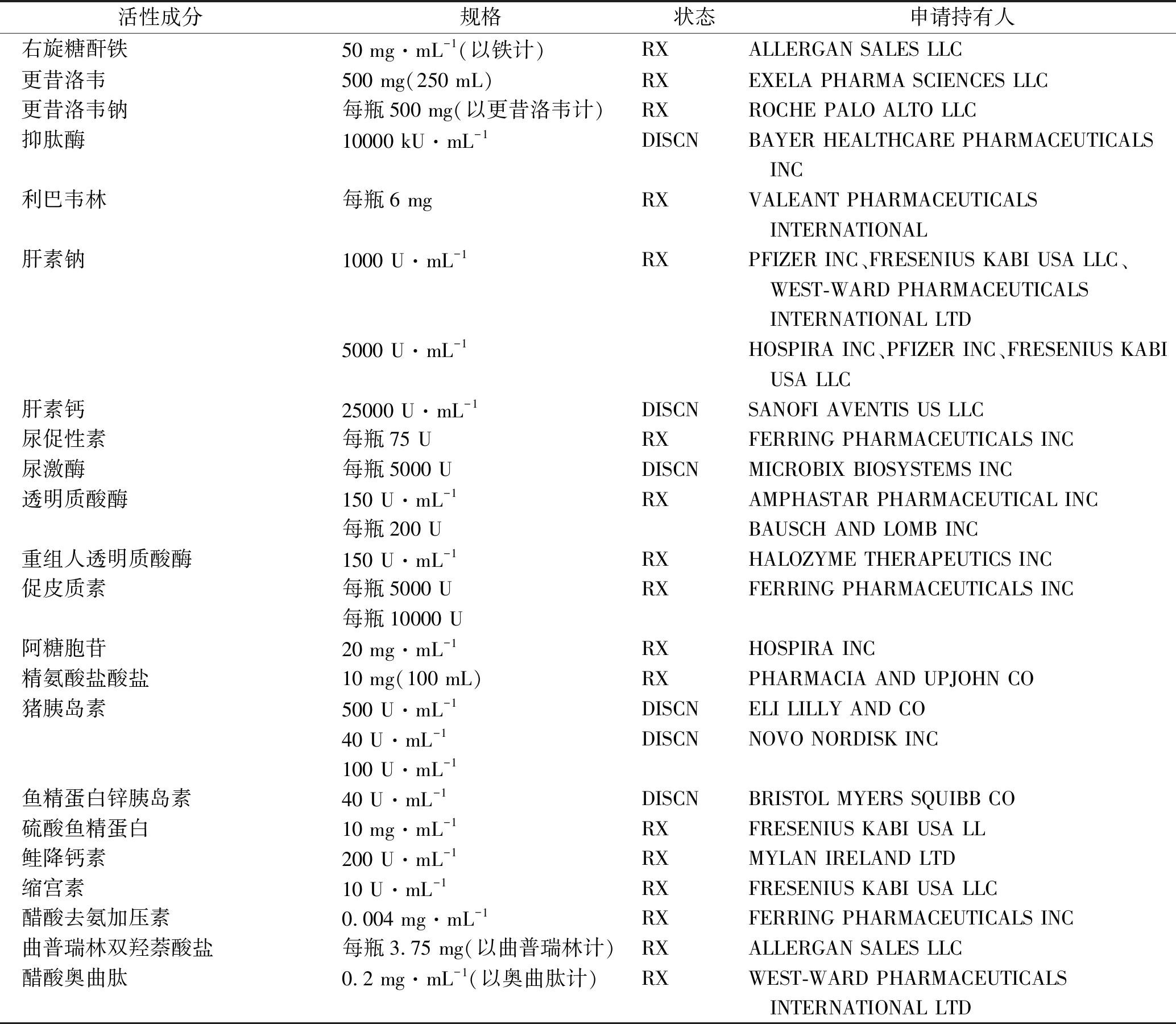
表1 FDA橙皮书上收载的生化药注射剂品种
RX表示处方药;DISCN表示停用药物
RX, prescription drug products; DISCN, discontinued drug products
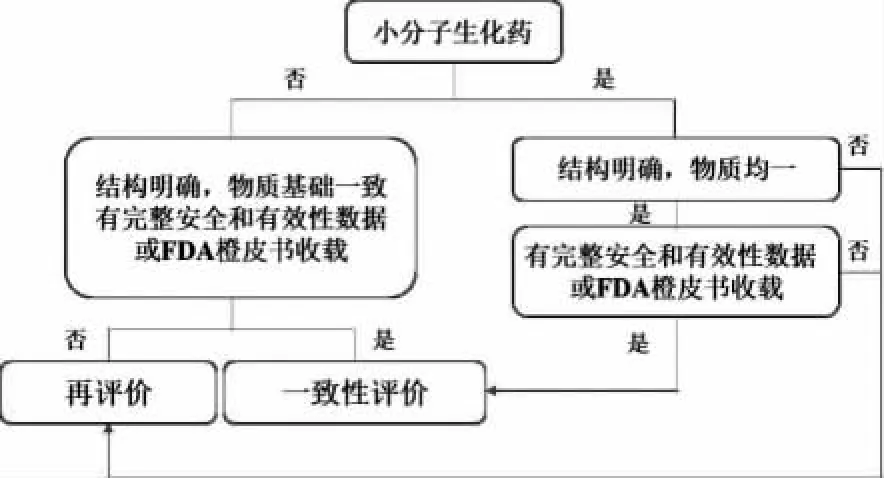
图1 生化药注射剂的评价策略
Fig.1Evaluationstrategyofbiochemicalinjection
3 生化药注射剂质量评价的药学研究要点
根据《多组分生化药注射剂基本技术要求(试行)》和《GMP生化药品附录》的要求进行归纳总结,生化药注射剂的药学研究要点主要有以下4点。
3.1剂型及规格的合理性 剂型的选择主要考虑药物的理化性质、稳定性和生物学特性,以及临床治疗的需要和临床用药的顺应性。此外,还要考虑工业化生产的可行性等因素。规格则是根据临床研究确定的用法用量,从方便临床用药、满足临床用药需要的角度设定,同时随着临床进一步的研究结果进行必要的调整。对于FDA橙皮书收载的生化药注射剂品种,应注意与橙皮书标识的参比制剂(reference standard,RS)保持一致。
3.2制备工艺研究 要确保与病毒传染有关的动物来源的生化药品的安全和质量,仅靠成品的质量标准不能完全控制此类产品的质量,必须从源头开始进行全程的严格控制,将原材料、中间体的质控作为质量标准的一部分。需要采用组合的方法,包括原材料的筛选、检疫和检测,提取纯化和病毒灭活(去除)工艺的验证,以及GMP的过程控制。在制剂处方及制备工艺研究中,因为生化药多存在性质不稳定、加热易破坏的问题,重点应确保生化药物理化学稳定性和生物活性。
3.3病毒灭活/去除工艺验证 生化药原材料来源于不同动物组织或体液,必须确立在生产工艺中包含能够有效灭活/去除病毒的特定工艺步骤,以保证产品的病毒安全性。病毒灭活/去除工艺验证主要应考虑生产工艺中是否含有有效的病毒灭活或去除生产工艺,至少应包含从机制上可以互补的两种有效工艺步骤,主要方法包括巴斯德消毒法、低pH孵放法、干热灭活法、有机溶剂处理法和膜过滤法等。
3.4质量研究与控制的技术要点及方法展望 生化药是按化学药管理的一类生物药物,其特点是相对分子质量大、不均一、易受生产工艺影响,可能包含的组分种类较多,如氨基酸、微量元素、短肽、蛋白质、酶、多糖、核酸及降解产物等,目前无法用一种手段来控制其质量,必须用组合技术手段来进行质量标准研究[5]。
常规化学药注射剂检验项目如pH值、澄清度与颜色、干燥失重、水分、细菌内毒素、热原、无菌、异常毒性、降压物质、不溶性微粒等同样适用于生化药注射剂的质量控制。此外,还有一些应用较多的新型技术手段,包括色谱、质谱、免疫化学、分子生物学、生物测定等技术,需要根据生化药所含组分的特征,采用合适的分析方法进行质量研究和控制[5]。
3.4.1动物源性原材料的种属鉴别 种属鉴别属于分子生物学技术,通常有常规聚合酶链反应(PCR)法、实时荧光PCR法和核酸测序法等。
常规PCR法是采用PCR技术对不同动物基因组中的特征性片段进行扩增,并进一步采用限制性内切酶酶切方法对扩增产物的序列进行确证,从而进行种属鉴别。实时荧光PCR法是对不同动物基因组中的特征性片段扩增的循环阈值(Ct值)进行测定,从而判定样品的种属来源。与常规PCR相比,实时荧光PCR法灵敏度高,操作简便,检测通量大,但仪器及试剂成本较高。笔者所在实验室目前已将常规PCR法和实时荧光PCR法应用到牛脾匀浆、小牛血清、垂体后叶、玻璃酸酶、肝水解肽、肝素钠等生化药原材料的种属鉴别[6-8]。DNA条形码分子鉴定法(也是核酸测序法)是利用基因组中一段公认的、相对较短的DNA序列来进行物种鉴定的一种分子生物学技术。主要用于中药饮片鉴别,生化药原材料鉴别还在验证。
3.4.2糖蛋白的糖基化分析 目前已上市的蛋白药物中三分之二以上是糖蛋白,如卵泡刺激素等。蛋白的糖基化与其功能密切相关,有必要对蛋白的糖基化进行分析。欧美药典收载的糖蛋白,在结构式描述中不仅给出氨基酸序列,而是将糖基化位点和糖链结构标出。因此,糖蛋白分析在表征生化药的结构上具有重要意义。糖蛋白药物的糖基化分析可分为完整蛋白水平、游离糖链水平、糖肽水平和单糖水平,使用的技术主要有色谱技术、质谱技术和毛细管电泳技术等[9]。笔者所在实验室采用色谱和质谱联用技术对尿促卵泡素的糖链结构进行分析[10],并完成我国第一个将糖蛋白结构中主要糖基化位点和糖链结构订入药品质量标准的制订工作。
3.4.3分子量与分子量分布测定 分子量与分子量分布是蛋白质、多糖类药物的重要质控指标之一,最常用的检测方法是分子排阻色谱法,测定的是相对分子质量,采用不同的分子量对照品及色谱柱会对测定结果产生影响。凝胶色谱联合多角度激光光散射检测法是近年来兴起的生物大分子分子量测定技术,无需分子量对照品的校正,完全不依赖于未知样品与标样结构及性质的相似性,可用于绝对分子量与分子量分布的表征,是目前各种测定分子量方法中最接近真值的方法,已陆续应用于羟乙基淀粉、琥珀酰明胶和玻璃酸钠注射液等多个生化药注射剂的测定并订入质量标准[11-12]。
3.4.4多肽的杂质分析 与经典小分子化学药相比,多肽在有关物质检查项目存在一定特殊性,由起始物料引入的杂质、工艺相关的杂质和降解产生的杂质等复杂多样,包含差向肽杂质、消旋肽杂质、链端杂质、侧链杂质、氧化/还原杂质、多聚体杂质等,这些杂质与多肽药物的疗效和不良反应紧密相关,必须加以控制。为实现有效检出各潜在杂质,通常需要采用多种不同原理的检测方法如反相高效液相色谱法(RP-HPLC)、离子交换高效液相色谱法、分子排阻高效液相色谱法等,也可采用液相色谱-质联用技术进行杂质分析[13]。
3.4.5氨基酸测定 氨基酸分析法主要用于肽的氨基酸组成、水解蛋白中氨基酸组成和复方氨基酸制剂中各组分的含量测定等,分析方法主要分为柱后衍生和柱前衍生两大类。其中常用的氨基酸柱前衍生化方法有异硫氰酸苯酯(phenyl isothiocyanate,PITC)衍生化、6-氨基喹啉-N-(羟基琥珀酰亚氨基)氨基甲酸酯(6-aminoquinolyl-N-hydroxy-succinimidel carbamate,AQC)衍生化、邻苯二醛(O-phthaldialdehyde,OPA)和9-芴甲基氯甲酸甲酯(9-fluorenylmethyl chloroformate,FMOC)衍生化、2,4-二硝基氟苯(2,4-dinitrofluorobenzene,DNFB)衍生化等,采用高效液相色谱仪测定;柱后衍生化方法主要有茚三酮衍生化,需用氨基酸分析仪测定[14]。欧美药典均收载,《中华人民共和国药典》2015年版还没有收载,但在生化药各论品种中应用广泛。
3.4.6免疫化学测定 免疫化学方法主要有免疫双扩散法、免疫电泳法、免疫印迹法和酶联免疫法等,这些方法已广泛应用于生化药的鉴别、纯度/杂质分析(包括病毒抗原或抗体的检测),同时也被用于效价测定、稳定性及其他质量属性的分析。欧美药典均收载了“免疫化学测定法”通则,《中华人民共和国药典》2015年版还未收载,目前正在起草。
3.4.7单核细胞激活测定 生化药来源复杂多样,工艺过程易受污染,必须进行热原检查。单核细胞激活测定法是热原家兔法的替代方法,属于生物测定中的安全性试验。传统热原检查方法有家兔法和细菌内毒素法,而单核细胞激活测定法是家兔法的替代方法,主要有人全血法、人外周血单核细胞法和单核细胞系法,2008年首次收载于《欧洲药典》(EP7.0),《中华人民共和国药典》《美国药典》和《日本药局方》目前均尚未收载该法。笔者所在实验室开发的单核细胞系法经过验证,可广泛用于疫苗、血液制品、单抗制剂和生化药品[15-16],在药品质量控制中具有应用前景。
3.4.8活性测定 除了理化和安全性检验项目外,活性测定也是评价生化药有效性的关键质量属性,是质量控制的重要组成部分。笔者实验室建立了多个体外活性测定方法,能有效对鹿瓜多肽注射液、注射用核糖核酸Ⅱ等生化药注射剂进行活性评价[17-18]。生物活性方法的建立必须进行方法学验证,但由于活性测定中剂量反应曲线遵循的数学模型与一般理化测定方法不同,如用得最多的一种模型是四参数回归模型,因此验证方法并不能采用理化测定方法常用的验证方法。目前《中华人民共和国药典》2015年版通则中对于生物活性尚没有给出具体的统计分析方法,不同标准中采用四参数logistic 模型计算供试品生物学活性的算法存在多种形式,通过不同算法获得的试验结果可能存在差异[19],因此生物活性和效价测定的验证方法需要进一步规范,笔者实验室正在拟订下一版《中华人民共和国药典》的“生物活性和效价测定方法验证指导原则”。
4 结束语
生化药注射剂由于来源特殊、成分复杂及其给药特点,是需要高度关注的风险较高的品种之一,有必要采用多种技术手段相结合的方式进行质量评价,以实现优胜劣汰,建立和健全结合起始原料和工艺过程的产品的整个生命周期的质量控制体系,全面提升我国生化药注射剂的质量水平。
猜你喜欢
中老年保健(2022年4期)2022-08-22
少儿美术(2019年1期)2019-12-14
小哥白尼(趣味科学)(2018年6期)2018-09-14
中学生数理化·高一版(2018年6期)2018-07-09
中成药(2018年5期)2018-06-06
中成药(2018年2期)2018-05-09
中国体育教练员(2017年3期)2018-01-19
中成药(2017年6期)2017-06-13
中成药(2017年3期)2017-05-17
