
一株临床耐药菌高通量测序数据的深度挖掘
2019-07-01 03:52傅行礼郑学明解冰刘永平高玉华吴晓蓉孔德华
中国老年学杂志 2019年12期
傅行礼 郑学明 解冰 刘永平 高玉华 吴晓蓉 孔德华
(1江苏大学医学部,江苏 镇江 212001;2镇江市丹徒区中医院;3南京市高淳人民医院)
临床上很多疾病的发生是由于致病菌的感染,而且很多致病菌已经进化为对多种抗生素有抗性的菌株〔1〕。传统的实验室培养方法可以得到细菌的形态学特征,从而知道该细菌的类型,抗生素敏感实验也能确定该菌株对哪些抗生素有抗性,这些结果对疾病的治疗提供了重要的信息。但要研究该菌株的基因组特征、来源及抗性机制等就必须进行全基因组测序。随着高通量测序技术的发展,细菌基因组测序已经同聚合酶链式反应(PCR) 一样方便和普及,并成为分子生物学研究的常规技术〔2〕。
高通量二代测序大大促进了生物信息学的发展,生物信息学家已经开发出了各种各样的软件工具对测序数据进行处理,但每一种软件都只解决数据处理某一个环节的一个具体问题〔3〕。怎么分析研究大量的测序原始数据,进而挖掘出有用的生物学信息,对于生物信息学家来说依然是一个巨大的挑战。而且如果能利用好现有的生物信息学工具对测序原始数据进行充分挖掘将会为实验研究提供重要的线索和方向,必将会大大加快研究的步伐。
本研究通过对一株临床耐药菌高通量测序数据的分析研究探讨信息提取、数据挖掘的流程。
1 材料和方法
1.1高通量测序数据的下载和质量控制 从美国国立生物技术信息中心(NCBI)的序列片段归档数据库(SRA)下载高通量全基因组测序(WGS)的原始数据(SRR2134675),测序样品是从成年女性血液中分离得到的Escherichia coli 菌株,测序方法是Illumina MiSeq双端(PE)测序。抗生素抗性实验表明该菌株对氨苄青霉素(ampicillin)、环丙沙星(ciprofloxacin)、磷霉素(fosfomycin)等多种抗生素有耐药性。下载的SRA文件用sratools 的fastq-dump 命令解压生成两个fastq文件。用Sickle 软件去掉平均质量值(Phred qualitiy)低于20(1% 测序错误)的序列,再去掉含有不确定碱基(N)数大于10%及长度不到30 bp的序列,从而得到高质量的数据进行下一步的分析。
1.2基因组denovo 组装和注释 用华大基因开发的SOAPdenovo软件对处理后的fastq 数据进行从头组装〔4〕。 选择不同的kmer 优化组装的结果,最后得到该菌株基因组组装的重叠群(Scaffolds),选择大于500 bp的重叠群序列进行下一步的分析。用细菌基因组注释软件Prokka对该菌株基因组草图进行注释〔5〕。
1.3COG分类分析 从NCBI下载微生物蛋白质相邻类的聚簇(COG)数据,建立本地blast数据库,用该菌株预测的所有蛋白质序列(fasta格式)逐一和本地COG 数据库中的蛋白质序列进行blastp 比对(E 值≤10-10,同一性≥40%,覆盖长度≥80%),选择匹配分数最高蛋白质的COG 分类注释查询蛋白,统计得到该基因组编码蛋白质的COG功能分类〔6〕。
1.4抗生素抗性基因分析 将该菌株基因组注释的蛋白质序列上传到抗生素抗性数据库(ARDB),使用blastp 程序和数据库中的抗性蛋白进行序列比对(E 值≤10-7,同一性≥40%,覆盖长度≥80%),根据同源性分析该菌株基因组中的抗性基因及抗性类型〔7〕。
1.5测序短序列(Reads)回帖(Read Mapping)和突变检出(Variation calling) 用多位点序列分型(MLST)软件MLST2.0分析该菌株基因组组装的Scaffolds序列,从而得到该菌株的分子分型〔8〕。通过分子分型找到和该菌株亲缘关系非常近的菌株作为参考基因组,用BWA( Burrows-Wheeler-Alignment Tool)软件把质控后的Reads Read Mapping到参考基因组上得到SAM(Sequence Alignment/Map) 文件,然后用SAMtools软件进行SAM-BAM格式转换及排序(Sorting),再用GATK软件对INDEL附近的Reads进行局部的重新比对,从而将比对的错误率降到最低,最后用BCFtools 软件检出突变(Variation Calling):单核苷酸多态性(SNPs)和插入缺失突变(INDELs)〔9~12〕。过滤掉等位基因频率低于75%及测序深度(Read-Depth)小于5的突变位点,生成包含高可信度突变位点信息的VCF(Variant Call Format)文件。
1.6突变位点的分析 从NCBI 下载大肠杆菌T131菌株EC958基因组的序列文件(fasta)及基因组注释文件(GFF3),用snpEff 软件建立该基因组的注释数据库〔13〕。 用snpEff软件对上一步BCFtools软件生成的VCF 文件进行注释:突变位点所在基因,突变对基因表达的影响,突变的危害程度等。
2 结 果
2.1基因组的一般特征分析 下载的全基因组测序数据共包含2 359 983对测序片段Reads,共1.1 G个碱基的序列数据。FastQC软件分析发现测序数据不含接头(Adaptor)和条形码(Barcode)序列,但其中一个文件的测序数据的3′末端序列平均质量不高,质量差异大。经过Sickle 软件质控后得到2 290 048 对高质量的测序片段。 SOAPdenovo 软件从头组装该菌株基因组共产生584 个重叠群,N50为 230 351 bp,GC含量为50.80%,预测基因组大小为5 278 663 bp。 组装时kmer 的选择会大大影响组装的质量,最好的办法就是测试不同的kmer,然后看N50的大小,选择N50 最高的组装结果。测序的数据量和测序质量是决定基因组组装好坏的最根本的因素,在测序数据量一定的条件下,选择高质量的序列进行组装会大大提高N50值。
Prokka 软件对组装的重叠群序列(Fasta文件)进行分析,结果表明该菌株基因组共有4 861个编码蛋白质的开发阅读框(ORFs)。进一步对这些蛋白质进行COG 分类分析,结果见图1。该菌株基因组编码的蛋白质中最多的是糖转运与代谢蛋白质(Carbohydrate Transport and Metabolism),467个ORFs;其次是氨基酸转运与代谢蛋白质(Amino Acid Transport and Metabolism),423个 ORFs及一般功能预测蛋白质(General Function Prediction Only),351个 ORFs。通过对基因组的组装和分析,可以得到该菌株基因组的整体特征,但由于基因组的复杂性和二代测序数据读长的限制,无法得到该菌株基因组的精细图,那些没有覆盖的序列(gap)大部分是基因组中的重复序列,不属于蛋白质编码区和基因表达调控区。
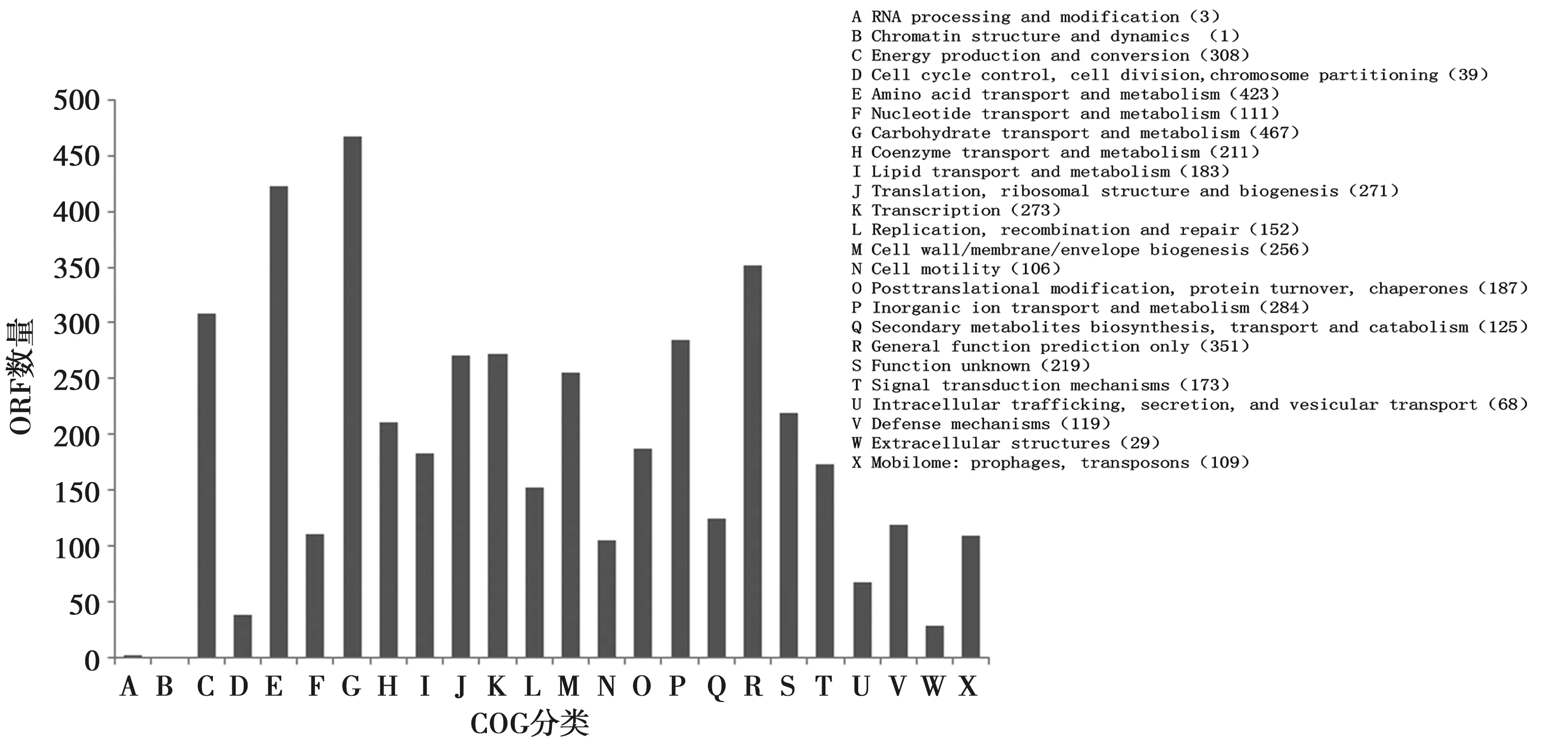
图1 预测蛋白质的COG 分类图
2.2抗生素抗性基因分析结果 抗生素抗性基因数据库ARDB预测出该菌株基因组中含有26 种抗性基因,见表1。该菌株基因组编码的蛋白质GHCCNJDC_03951 属于mdtg 抗性基因类型,GHCCNJDC_03939 属于mdth 抗性基因类型,这和该菌株对磷霉素有抗性的实验结果一致;GHCCNJDC_04736属于bl2be_ctxm 类型,这一预测结果也和该菌株对氨苄青霉素有抗性的实验结果一致。此外还有属于acra 抗性基因类型的GHCCNJDC_00668,属于acrb抗性基因类型的GHCCNJDC_00669等。在这些预测的抗生素抗性基因中,除了ACD 类β-内酰胺酶外,绝大多数属于多药耐药外排泵 (Multidrug Resistance Efflux Pump)。

表1 抗生素抗性基因预测结果

续表1 抗生素抗性基因预测结果
2.3菌株的鉴定和比较基因组学研究 通过从头组装,虽然得到了该菌株基因组的草图,但仍然无法回答该菌株是从什么菌株进化而来,其基因组发生了哪些突变从而产生耐药性。首先,用分子分型软件MLST对该菌株的重叠群序列进行分析,结果显示该菌株为ST131。MLST软件是根据七个保守的基因序列(adk,fumC,gyrB,icd,mdh,purA及recA)把大肠杆菌分成不同的序列类型(ST),相同ST的菌株之间基因组差异小,亲缘关系近。 由于大肠杆菌不同菌株基因组的差异,如果不进行分子分型找到参考基因组而直接用大肠杆菌MG1655菌株作为参考基因组,将会找到几十万乃至上百万的突变位点,而这些突变位点绝大多数都是由于错误的比对导致的。
通过分子分型,笔者知道了该菌株的进化地位,并找到了进行比较基因组研究的参考基因组。接下来,把该菌株Reads Mapping到大肠杆菌T131菌株EC958 基因组上,从而找到该菌株和参考基因组不一致的突变序列。BWA 回帖结果显示该菌株和大肠杆菌EC958 菌株基因组有很高的同源性,平均测序深度(Depth)为97倍,覆盖度几乎百分之百(仅仅100个位点的测序深度为零)。突变检出(Variation Calling)共找到1 562个突变位点,包括1 536个高可信度SNPs和 26个插入缺失突变(INDELs)。
2.4突变位点snpEff 注释结果 为了进一步分析这些突变,采用snpEff 软件结合参考基因组的注释数据对突变数据(VCF) 进行注释。注释结果如表2所示。 绝大多数突变属于SNP(1 536个),少数属于INDEL(26个)。SNP突变中位于基因编码区的有1 187个,位于基因表达调控区349个。位于编码区的SNP突变中,859个是不改变蛋白质序列的同义突变(synonymous mutation),错义突变(missense mutation)有326个, 此外还有2个终止突变(stop_gained mutation)。两个终止突变将使相应的蛋白质翻译提前终止:OmpF 蛋白(WP_000977908.1)基因编码区第250位的鸟嘌呤转换成胸腺嘧啶 (c.250G>T) 从而导致该蛋白84位的谷氨酸变成终止位点(p.Glu84*);Ves蛋白(WP_000455604.1)基因编码区的突变(c.460C>T),导致终止密码子的产生(p.Gln154*), 从而使该蛋白的翻译提前终止。大多数插入删除(INDELs)突变都会导致读码框的改变,以致不能产生正常的蛋白质。深入分析这些突变位点将有可能发现导致抗生素抗性的基因和突变。

表2 snpEff注释结果
3 讨 论
随着第二代高通量测序技术的发展,数据库中各种生物序列数据呈爆炸式增长,这些生物大数据中蕴含丰富的信息,怎么去分析挖掘这些信息是目前面临的急需解决的问题。本研究通过对一株临床多药抗性菌株的高通量二代测序原始数据进行充分挖掘,最终找到可能的抗药性突变位点,为后续的研究提供了重要的线索。
通过对该菌株基因组测序数据的处理和从头组装,我们得到了该菌株的基因组草图。由于基因组中很难组装的部分都位于高度重复区域,这些区域一般不是蛋白编码基因或表达调控序列,所以根据基因组草图,我们能预测出ORFs并进行多药抗性基因分析、COG分析等。再根据该菌株的基因组草图进行分子分型,从而确定了该菌株的进化地位及参考基因组。通过把测序片段回帖到参考基因组、突变检出、突变注释等进行一系列的生物信息学分析,最终得到了该菌株和参考基因组不一致的所有突变位点,这些突变将为研究该菌株的耐药机制提供重要的线索。
随着越来越多的细菌全基因组精细图谱的产生,分子分型将越来越精准,我们将更准确地确定菌株的进化地位,检出突变,为实验科学提供更有价值的线索。随着测序数据的爆炸式增长,生物信息学家需要开发出能大规模分析处理二代测序原始数据的优秀挖掘算法和分析流程,这将使我们能从临床致病菌的测序数据中挖掘出更多更有价值的信息。
猜你喜欢
分子催化(2022年1期)2022-11-02
当代水产(2022年7期)2022-09-20
河北科技师范学院学报(2022年2期)2022-08-26
军事文摘(2022年16期)2022-08-24
中国饲料(2021年17期)2021-11-02
今日农业(2021年11期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
空间科学学报(2021年1期)2021-05-22
中国生殖健康(2020年4期)2020-12-09
