
高糖诱导的大鼠原代心肌细胞损伤对程序性坏死的影响及其机制*
2019-06-25 02:27方婷婷曹瑞平叶红伟马善峰
中国应用生理学杂志 2019年2期
方婷婷,曹瑞平,叶红伟,马善峰,高 琴
(蚌埠医学院生理学教研室,安徽 蚌埠 233030)
糖尿病(diabetes mellitus,DM)是世界上最常见的非传染性慢性疾病之一,随病程延长,往往伴有多脏器并发症。高血糖引起的心肌损伤是糖尿病最严重的并发症之一,表现为心肌氧化应激损伤、炎症反应、细胞死亡,最终导致心力衰竭。细胞死亡是生命的基本组成部分,包括凋亡、自噬、坏死等多种形式。随着研究的深入,Degterev A团队于2005年在小鼠脑缺血损伤中第一次报道另一种可调控性的细胞坏死方式——程序性坏死(necroptosis)[1]。Necroptosis也称为坏死性凋亡,是一种由激酶的激活引起的细胞死亡方式,兼具凋亡和坏死特征,又不同于这两种细胞死亡方式。与经典的细胞凋亡相比,necroptosis虽然可调控,但不形成凋亡小体,染色质不聚集,不依赖半胱氨酸天冬蛋白酶(caspase),但伴有活性氧的增加,发生炎症反应,可被Nec-1特异性抑制,而不受凋亡抑制剂(如z -VAD)的影响;与经典的坏死相比,necroptosis具有坏死样的形态学变化,出现细胞膜/器破裂、核裂解等,但受多种基因调控,主要通过受体相互作用蛋白激酶1(receptor interacting serine/threonine protein kinase 1,RIP1)、受体相互作用蛋白激酶3(receptor interacting serine/threonine protein kinase 3,RIP3)与混合谱系激酶结构域样蛋白(mixed lineage kinase domain-like protein,MLKL)形成RIP1-RIP3-MLKL轴,称为“坏死复合物”。RIP3与RIP1通过各自的RIP同源结构相互作用,RIP3是RIP1的下游调节因子,RIP3诱导MLKL寡聚化转位至细胞质膜,与磷脂酰肌醇相互作用触发细胞膜通透性增强,并破坏细胞导致细胞死亡[2-5]。
近年来,程序性坏死作为一种受调控的细胞坏死形式,引起人们的广泛关注[6]。各种损伤性刺激如氧化应激、缺血、炎症等均可诱发程序性坏死的发生,与心肌梗死、中风、缺血/再灌注损伤等疾病密切相关,有效抑制necroptosis的发生可直接抑制炎症反应,减轻疾病损伤。心肌损伤作为糖尿病最常见的并发症之一,其发病机制尚不完全明确,深入探讨其发生机制具有重要的临床应用价值。值得注意的是,高糖引起的心肌损伤中伴有氧化应激、炎症等的发生,那么高糖是否诱导心肌细胞发生程序性坏死?抑制程序性坏死是否可减轻高糖引起的心肌损伤和炎症反应?以上问题值得我们深入探讨。因此本研究首先观察了程序性坏死在高糖诱导的原代心肌细胞损伤中变化,并探讨其可能机制,为临床预防及治疗糖尿病心肌损伤提供理论基础。
1 材料与方法
1.1 实验动物和主要试剂材料、仪器设备
SPF级1~3 d SD乳鼠,由蚌埠医学院实验动物中心提供,合格证号:SCXK(苏 2017-0001)。Nec-1(necrostatin-1,RIP1的特异性抑制剂)、二氢乙啶(dihydroethidium,DHE)、MTT均购于美国Sigma公司,TNF-α、IL-6及IL-1β试剂盒均购于达科为生物技术有限公司,总RNA的提取试剂Trizol购自美国Invitrogen公司,RNA逆转录试剂盒购自美国Thermo Fisher公司,Real-time PCR试剂盒购于日本Takara Clontech公司,各引物由上海生工生物公司合成,RIP1引物:上游序列5'- AGG TAC AGG AGT TTG GTA TGG GC-3',下游序列 5'- GGT GGT GCC AAG GAG ATG TAT G-3',扩增产物长度约为123 bp;RIP3引物:上游序列 5'- TAG TTT ATG AAA TGC TGG ACC GC-3',下游序列 5'- GCC AAG GTG TCA GAT GAT GTC C-3',扩增产物长度约为145 bp;MLKL引物:上游序列 5'- GCC ACT GGA AAG ATC CCG TT-3',下游序列 5'- CAA CAA CTC GGG GCA ATC CT-3',扩增产物长度约108 bp;以GAPDH为内参,上游序列 5'- ACA GCA ACA GGG TGG TGG AC-3',下游序列5'-TTT GAG GGT GCA GCG AAC TT -3',扩增产物长度约为255 bp。兔抗大鼠RIP1抗体、RIP3抗体均购自美国Abcam公司,兔抗大鼠MLKL抗体、GAPDH抗体分别购于英国biorbyt公司、上海爱必信生物公司。
倒置荧光显微镜(Cat No.1X71)购自日本Olympus公司。Real-time PCR仪(Applied Biosystems® StepOnePlus)为美国Applied Biosystems公司产品。酶标仪(Cat No.Synergy 2)为美国Bio-Tek公司。小型转印槽(Mini Trans-Blot®)、垂直电泳槽(Mini-PROTEAN® Tetra Cell和成像仪(ChemiDocTMXRS+ System)为美国BIO-RAD公司产品。
1.2 大鼠原代心肌细胞培养
取1~3 d的乳鼠心尖组织,放于预冷的HBSS液中挤压出血,清洗,眼科剪将心脏组织块剪成1 mm3块状,加入终浓度为10 μg/ml的DNA酶I、0.08%的II型胶原酶、0.07%的胰蛋白酶混合液进行消化,37℃、5%CO2培养箱静置7~10 min,弃上清。重复消化步骤约5~7次,直至组织块蓬松。加入低糖完全培养基(4.9 mmol/L葡萄糖)终止消化,轻柔吹散,静置,待组织沉淀后吸取混浊上清液入一离心管中。重复步骤直至组织中的液体清亮,收集的混浊上清液1 000 r/min、离心6 min,弃上清,获取细胞沉淀。低糖完全培养基重悬细胞沉淀,种入培养皿中,于培养箱培养90 min后,小心吸取培养液1 000 r/min、离心6 min,获取细胞沉淀,F12完全培养基将其重悬,加入5-Brdu(终浓度为0.1 mmol/L,抑制混杂的成纤维细胞),种入培养皿中于培养箱培养。显微镜下观察心肌细胞,培养至3~4 d,心肌细胞相互接触交织,出现同步化搏动。
1.3 实验分组
F12完全培养基培养心肌细胞,干预前予以无血清培养基处理24 h,使其同步化。实验随机分为4组:(1)正常对照组(normal control group,Control):心肌细胞在葡萄糖浓度为5.5 mmol/L的完全培养基中培养48 h;(2)高糖组(high glucose,HG):葡萄糖浓度为30 mmol/L的完全培养基培养48 h[7];(3)高糖+Nec-1组(HG+Nec-1):将RIP1的特异性抑制剂Nec-1加入葡萄糖浓度为30 mmol/L的完全培养基中,其终浓度为100 μmol/L[8],培养48 h;(4)高渗组(hypertonic pressure group,HPG):含 5.5 mmol/L葡萄糖+24.5 mmol/L甘露醇的完全培养基,培养48 h。
1.4 MTT检测各处理组细胞活力
心肌细胞计数,每孔1.0×104个细胞种在96孔板于5% CO2培养箱培养,每组设置5个孔,四周孔加入PBS液,实验重复3次。实验处理前予以无血清培养基同步化处理24 h。实验处理48 h后各孔加入20 μl MTT溶液(工作浓度:0.5 mg/ml),37℃孵育4 h,生成深紫色结晶,小心去除液体,各孔加入150 μl DMSO,置于摇床上摇15~30 min,使结晶完全溶解,上机,检测波长在490 nm处的OD值。
1.5 DHE检测不同处理组心肌细胞氧化应激水平
每孔2×105个细胞种于6孔板,于5% CO2培养箱培养,实验处理前予以无血清培养基同步化处理24 h,实验干预处理48 h后,PBS清洗3次,每次3 min,按照说明书各孔加入含有DHE的F12完全培养基(DHE工作浓度:10 μmol/L),避光,37℃孵育30 min,PBS清洗3次,每次5 min,然后4%多聚甲醛固定10 min,倒置荧光显微镜下拍照。使用ImageJ1.48v软件对各组图片进行量化分析。
1.6 ELISA法检测不同处理组心肌细胞TNF-α、IL-6及IL-1β水平
每孔1×106个细胞种于培养皿于5% CO2培养箱培养,实验处理前予以无血清培养基同步化处理24 h,实验干预处理48 h后,收集上层培养液备用,准备5批次样品,按照试剂盒说明书操作:加样,洗板,加检测抗体,洗板,加酶,洗板,显色,终止反应,读板:终止后 10 min内,使用检测波长450 nm、校正波长610~630 nm 双波长同时读板。
1.7 Real-time PCR检测不同处理组心肌细胞RIP1、RIP3、MLKL mRNA水平变化
Trizol提取心肌细胞总 RNA,酶标仪测总RNA的纯度和浓度,取3 μg总RNA 作模板,按照逆转录试剂盒说明书合成cDNA,按照Real-time PCR 试剂盒说明书对应的Applied Biosystems®Step One Plus 仪器,取2 μl cDNA(50 ng/μl)为模板,加入上下游引物各0.6 μl(终浓度0.3 μmol/L),SYBR® Premix DimerEraser(2×)10 μl,ROX Reference Dye 0.4 μl,加入无酶水使总体积为20 μl,进行上机扩增。扩增条件:(1)预变性95℃、30 s,(2)变性95℃、5 s,(3)退火60℃、30 s,(4)延伸72℃、34 s,(2)(3)(4)40个循环,(5)溶解曲线条件由仪器自动生成。用内参GAPDH进行校正,以正常组作为对照,计算2-ΔΔCt值为各处理样品基因的相对表达量,进行数据分析。
1.8 Western blot检测不同处理组心肌细胞RIP1、RIP3、MLKL蛋白水平变化
收集不同批次培养的心肌细胞沉淀,加入细胞裂解液(RIPA)和PMSF(终浓度为1 mmol/L),提取细胞总蛋白,按照试剂盒说明书检测蛋白浓度。获取的蛋白按照40 μg对应的体积加样,进行SDS-PAGE(聚丙烯酰胺凝胶)电泳(10%分离胶90 V,5%浓缩胶60 V)。然后转膜,200 mA、120 min。5%的脱脂牛奶(脱脂奶粉+TBST液)常温封闭150 min,加入RIP1抗体(1∶500)、RIP3抗体(1∶500)、MLKL抗体(1∶800)、GAPDH抗体(1∶ 6 000),4℃孵育过夜。次日洗膜后,加二抗IgG(1∶ 7 000),孵育60 min,TBST洗膜后,加入显影液,凝胶成像系统曝光。以GAPDH为内参,凝胶成像系统进行条带灰度值扫描,分别算出RIP1、RIP3、MLKL与GAPDH的灰度值比值。
1.9 统计学处理
2 结果
2.1 不同处理组心肌细胞活力的比较
MTT结果反映心肌细胞活力变化。结果显示,正常对照组OD值为(0.601±0.033)。与正常对照组相比,高糖组OD值为(0.368±0.010),心肌细胞活力明显下降(P<0.01);与高糖组相比,高糖+Nec-1组OD值为(0.528±0.045),心肌细胞活力明显升高(P<0.01)。与正常对照组相比,高渗组OD值(0.567±0.089),心肌细胞活力无显著差异,提示高渗环境对心肌细胞活力无明显影响,因此在后续实验中未设高渗实验组。
2.2 不同处理组心肌细胞氧化应激的比较
DHE为活性氧簇的荧光探针,其可穿透活细胞膜进入胞内,氧化活性氧簇,产生氧化乙啶,并与染色体DNA结合,发出红色荧光。根据红色荧光的强度,可判断各组心肌细胞氧化应激变化。正常对照组平均荧光强度为(0.0130±0.0004);高糖组平均荧光强度为(0.0336±0.0003),与正常对照组相比,荧光强度明显增强(P<0.01),提示氧化应激水平增高;高糖+Nec-1组平均荧光强度为(0.0204± 0.0010),与高糖组相比,荧光强度明显减弱(P<0.01),提示氧化应激水平降低(图1)。

2.3 不同处理组心肌细胞TNF-α、IL-6及 IL-1β水平的比较
与正常对照组相比,高糖组TNF-α、IL-6及IL-1β水平升高明显(P<0.01);与高糖组相比,高糖+Nec-1组TNF-α、IL-6及IL-1β水平明显降低(P<0.01,表1)。
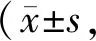
Tab. 1 Levels of TNF-α,IL-6 and IL-1β in different groups n=5)
HG:High glucose group;HG+Nec-1:High glucose+necrostatin-1 group;TNF-α:Tumor necrosis factor-α;IL-6:Interleukin-6;IL-1β:Interleukin-1β
**P<0.01vscontrol group;##P<0.01vsHG group
2.4 不同处理组心肌细胞中RIP1、RIP3、MLKL mRNA及蛋白表达的比较
与正常对照组相比,高糖组中RIP1、RIP3、MLKL mRNA及蛋白表达均升高(P<0.01);与高糖组相比,高糖+Nec-1组RIP1、RIP3、MLKL mRNA及蛋白表达均降低(P<0.05~0.01,表2,3、图2)。
Tab. 2 Expressions of RIP1,RIP3 and MLKL at mRNA level in cardiomyocytes in different groups n=5)
RIP1:Receptor interacting serine/threonine protein kinase 1;RIP3:Receptor interacting serine/threonine protein kinase 3;MLKL:Mixed lineage kinase domain-like protein
**P<0.01vscontrol group;#P<0.05,##P<0.01vsHG group
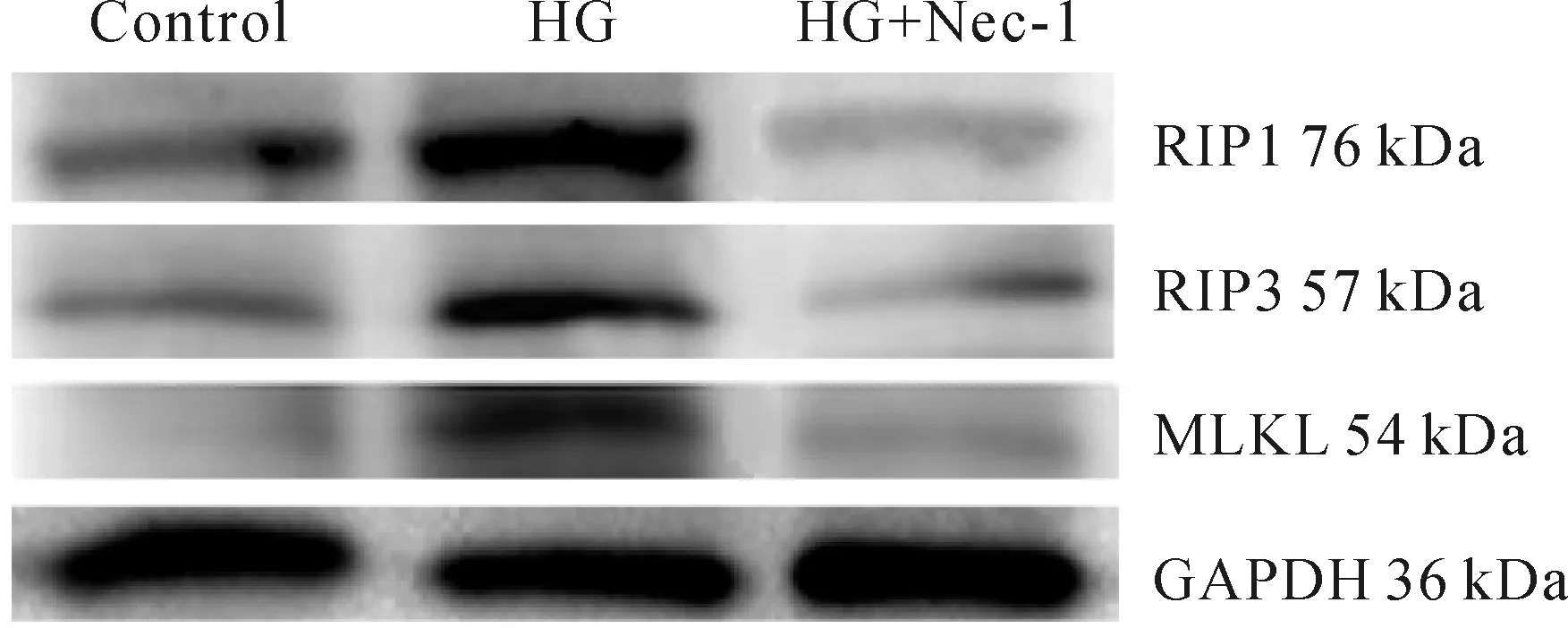
RIP:Receptor interacting serine/threonine protein kinase;MLKL:Mixed lineage kinase domain-like protein
Tab. 3 Changes of RIP1,RIP3 and MLKL protein expressions in cardiomyocytes in different groups n=4)
*P<0.05,**P<0.01vscontrol group;#P<0.05,##P<0.01vsHG group
3 讨论
糖尿病心肌损伤是糖尿病最严重的心血管并发症之一,严重影响人类的生活质量。高血糖引起心肌氧化应激、炎症反应、心肌纤维化、细胞死亡等,最终导致心力衰竭[9,10]。我们通过检测观察到高糖培养的心肌细胞活力明显下降,高渗对心肌细胞活力无明显影响,提示高糖本身可模拟糖尿病高血糖状态诱导心肌细胞损伤。
慢性炎症和氧化应激在高血糖引起的心血管并发症中发挥重要作用。高血糖引起炎症因子和氧自由基生成增加,导致细胞功能损害[11,12]。文献报道,TNF-α刺激程序性坏死发生,且可刺激炎症反应[13]。多种信号如脂多糖、钙离子载体、细胞因子等和一些病理状态如外伤、细菌和病毒感染、自身免疫性疾病、炎症等均能上调IL-6水平。IL-1β通常在正常细胞中分泌较少,但在应对刺激如炎症物质、感染、细菌性内毒素时,IL-1β水平明显提高。疾病进展期间减缓炎症和氧化应激可能是治疗高血糖引起心血管疾病的潜在治疗手段之一。本实验观察到,与正常对照组相比,高糖处理的心肌细胞ROS产生增加,TNF-α、IL-6和IL-1β水平明显升高,提示高糖诱导心肌细胞氧化应激及炎症反应的发生。但高糖如何引起氧化应激和炎症反应值得探讨。
长期以来细胞坏死都被认为是一种被动且不可调控的过程。近年研究表明,细胞坏死也是受到精密调控的,程序性坏死作为规律性细胞死亡形式之一与许多疾病的病理和器质损伤密切相关,可能涉及细胞内感染的防御过程[14]。文献报道,心肌细胞缺氧/复氧时,RIP1、RIP3蛋白表达水平显著升高,D-半乳糖致衰老小鼠模型中,抑制RIP1通过抑制神经炎症反应改善脑损伤[15,16]。本研究中,我们观察到原代心肌细胞在高糖干预后,炎症反应发生的同时,程序性坏死关键介质RIP1、RIP3和MLKL的mRNA水平和蛋白表达均升高,提示高糖可诱导心肌细胞程序性坏死的发生;程序性坏死本身可能诱发氧化应激和炎症反应。
据报道,抑制程序性坏死的发生对体内外动物心肌损伤均发挥有益作用。RIP1-RIP3复合物的形成和磷酸化是necroptosis的关键性和特异性步骤,是necroptosis发生的标志[17,18]。Nec-1是RIP1选择性变构抑制剂,特异性抑制RIP1的激酶活性及RIP1和RIP3的相互作用[19],Nec-1在小鼠及猪心肌缺血/再灌注损伤中均可抑制程序性坏死,保护心肌免受损伤[20-22],Nec-1抑制程序性坏死途径减缓百草枯诱导的心脏收缩功能障碍[23],提示各种诱因诱发的心肌损伤中,抑制程序性坏死可发生保护作用。本研究中,高糖处理的心肌细胞中给予Nec-1抑制RIP1后,RIP1、RIP3和MLKL的mRNA水平和蛋白表达均降低的同时心肌细胞存活率增高,ROS的产生和炎症因子释放减少,进一步表明程序性坏死与心肌损伤中的氧化应激和炎症反应关系密切。抑制程序性坏死可减轻氧化应激及炎症反应,从而发挥心肌保护作用,其具体机制还有待进一步深入探究。
综上所述,在高糖诱导的心肌细胞损伤中伴有程序性坏死的发生。抑制程序性坏死可能通过减轻氧化应激及炎症反应而减弱高糖诱导的心肌损伤。如何有效对抗程序性坏死的发生可能是治疗糖尿病心肌损伤的重要途径之一。
猜你喜欢
眼科新进展(2022年12期)2022-12-29
世界科学技术-中医药现代化(2022年2期)2022-05-25
眼科新进展(2022年2期)2022-03-11
现代临床医学(2021年5期)2021-11-02
天津医科大学学报(2021年4期)2021-08-21
心肺血管病杂志(2020年5期)2021-01-14
意林·少年版(2019年11期)2019-06-30
科学之谜(2019年3期)2019-03-28
体育科学(2018年12期)2019-01-04
科学中国人(2016年9期)2016-11-04
