
氢卟啉,氯取代卟啉结构和吸收光谱性质的理论研究
2019-05-31 02:48诸葛霄李云志刘国魁夏其英
山东化工 2019年9期
诸葛霄,冷 霞,李云志,刘国魁,夏其英
(临沂大学 化学化工学院,山东 临沂 276000)
由于其天然存在的大环结构,卟啉及其衍生物表现出良好的光理化和氧化还原性能,在科学研究和工业应用等领域备受关注。卟啉类化合物广泛存在于生命内,如植物体内的叶绿素,人体内的血红素等。这激起许多研究聚焦于其光电方面的应用。卟啉及其衍生物在半导体、太阳能电池、光电功能材料等方面有重要的应用[1]。因此,研究卟啉类化合物的吸收光谱,分析影响其光性能的因素,对设计发展具有优良性能的含卟啉光电材料有重要的指导意义。
卟啉是在卟吩环上拥有取代基的一类大环化合物的总称,具有较强的可修饰性。实验和理论对卟啉类化合物的设计合成、分子结构及光谱性质的关注越来越多。Sánchez-Bojorge 等人[2]通过实验合成表征了系列四meso位取代卟啉化合物,结合理论计算发现不同的取代基对分子的几何构型没有明显的影响,但是可以改变电子结构和轨道能级。Kong等人[3]通过实验合成了Neo-N混杂卟啉及其氯代衍生物,发现两者有相似的分子结构和紫外吸收光谱性质。通过光谱实验和理论计算,Balanay等人[4]研究了溶剂和外加电解质对两种卟啉金属配合物的光电性能的影响。许多研究表明不同极性的溶剂是引起卟啉类化合物吸收光谱变化的一个重要因素[5-8]。
获得卟啉类化合物,可以对其进行分子内重排获得同分异构体,进行C-N位置互换得到N混杂卟啉以及在不同位置连接不同数量和类型的取代基团进行修饰。研究发现,卟啉类分子上的C-N重排[5-6,9],以及引入取代基改变分子原有的对称性[9]都会引起不同程度的轨道能级和光谱性质的改变。Ngaojampa等人[10]发现在meso位取代基中引入电子供体组分可以引起光谱的蓝移,Soret和B带的分裂。Bonnett等人实验研究表明卟啉环上引入氯原子对光谱具有明显的影响[11]。
虽然目前对氯取代卟啉类化合物的研究得到了实验的研究。但是,对于氯原子在不同位置对卟啉分子结构结构的影响,以及不同溶剂的影响,还缺乏系统的理论研究。本文将应用DFT和TDDFT理论,计算自由卟啉(FBP)、2-氯卟啉(2-Cl-FBP)、4-氯卟啉(4-Cl-FBP)和2'-氯卟啉(2'-Cl-FBP)四种卟啉类化合物的基态结构和吸收光谱,同时计算四者在气态和不同极性溶剂条件下的结构和吸收光谱,解释FBP、2-Cl-FBP、4-Cl-FBP和2'-Cl-FBP由于Cl原子位置的变化而引起的基态结构和吸收光谱的变化规律,以及苯、二氯甲烷和水三种不同极性溶剂对这四种化合物基态结构和吸收光谱的影响规律。
1 计算模型和方法
计算采用Gaussian 09程序包[12],在气态和不同溶剂中在M062X/6-31+G(d)水平上对FBP、2-Cl-FBP、-Cl-FBP和2'-Cl-FBP的基态几何结构进行优化及振动频率分析,其中FBP在D2h对称性下优化,2-Cl-FBP、-Cl-FBP和2'-Cl-FBP不加任何对称性限制,并且通过频率分析确定没有虚频。采用TD-M062X/6-311++G(d,p)方法在溶剂极化连续模型下计算气态及苯、二氯甲烷和水不同环境下的电子吸收光谱。采用GaussSum软件在半峰宽为1500 cm-1条件下产生电子吸收光谱。
2 结果与讨论
2.1 基态分子的几何结构分析
在气态条件下优化的卟啉(FBP)、2-氯卟啉(2-Cl-FBP)、4-氯卟啉(4-Cl-FBP)和2'-氯卟啉(2'-Cl-FBP)的几何结构见图1。图1表明,卟啉外环上的H原子被Cl原子取代之后对内环的N-N及H-H的距离没有产生很大的影响;Cl原子在卟啉环上位置不同时,对整个大环的键长没有显著的改变。外接C-Cl之间距离的最大差值为0.003 nm,内部H-H之间的距离没有改变,N-N之间的距离最大相差0.001 nm。其中FBP具有平面结构(D2h对称性)。由于Cl原子的引入,破坏了该对称性,但是2-Cl-FBP、4-Cl-FBP和2'-Cl-FBP也几乎是一个平面。

图1 FBP(a)、2-Cl-FBP(b)、4-Cl-FBP(c)和2'-Cl-FBP(d)的分子结构(键长单位为
2.2 分子的光谱和分析轨道分析
所有研究体系在气态和不同溶剂下的电子吸收光谱见图2。四个体系在气态下Soret带均出现较强的吸收峰,不同Cl的取代引起Soret的吸收峰位置(347 nm)均发生红移,分别为351nm(2-Cl-FBP)、354nm(4-Cl-FBP)、350nm(2'-Cl-FBP)。同时,不同Cl取代位置对Soret强度也有影响,但是没有明显的规律。

图2 FBP(3)、2-Cl-FBP(1)、4-Cl-FBP(4)和2'-Cl-FBP(2)四种分子在气态(a)、
苯(b)、二氯甲烷(c)和水(d)中模拟的吸收光谱
FBP在Q带有两个峰高相等的弱吸收峰,这主要是由于FBP具有比较高的对称性(D2h),Qx和Qy两个吸收跃迁的偶极大小相近,但是方向相反,因而吸收强度很低。不同位置氯原子的取代主要影响Q带的峰高,对峰位置的影响没有明显规律。FBP有两个强度类似的吸收峰,这主要是由对称性引起的。氯原子的引入打破了原有的对称性。从图2中发现,2-Cl-FBP和4-Cl-FBP都会使Qx的吸收峰变强,而Qy的吸收峰变弱。而且,2-Cl-FBP的影响比4-Cl-FBP的影响更强烈。2'-Cl-FBP则增强了Qy的吸收。

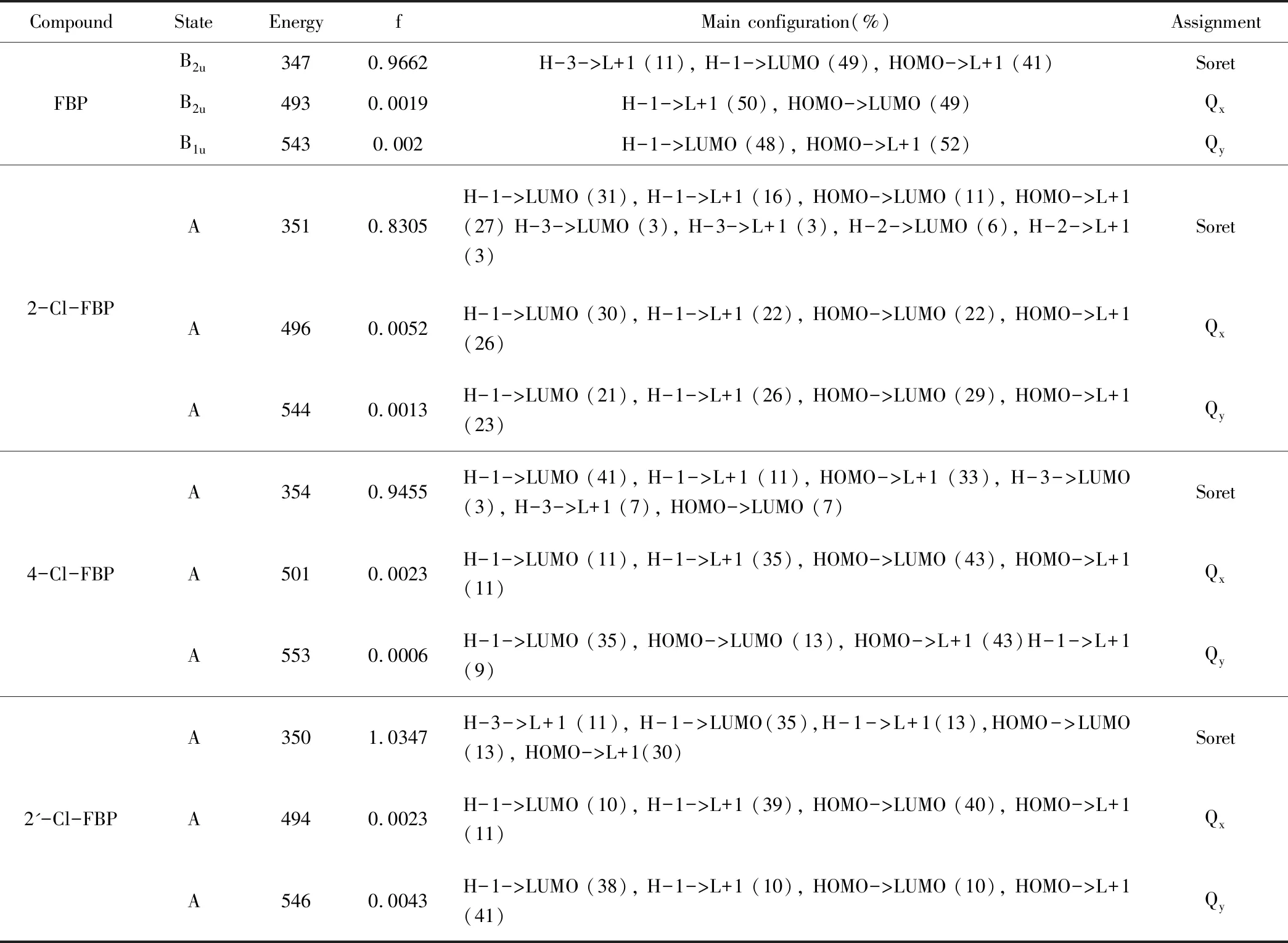
图3 FBP、2-Cl-FBP、4-Cl-FBP 和 2'-Cl-FBP的轨道分布
Gouterman用HOMO-1、HOMO、LUMO和LUMO+1来解释卟啉的吸收光谱。由表1可知,对于Soret带还有部分HOMO-3到LUMO+1的跃迁,需要说明的是这个比重是很小的(11%)。因此,我们说用四个轨道解释相对合理。
FBP的跃迁在组成上比较简单,Soret 的组成为H-3->L+1 (11), H-1->LUMO (49), HOMO->L+1 (41),Qx的组成为H-1->L+1 (50), HOMO->LUMO (49),Qy的组成为H-1->LUMO (48), HOMO->L+1 (52)。当体系中引入氯原子之后,每个光谱带的跃迁都变的更加复杂。比如FBP的Soret带由原来的3种组成,在引入氯原子后分别变成8种(2-Cl-FBP),6种(4-Cl-FBP),5种(2'-Cl-FBP)。对于Qx和Qy也有类似的现象。这说明氯原子的引入增加了轨道之间的相互作用,从而对吸收光谱造成影响。
四个体系轨道分布如图3。从图3中可以看出氯离子引入后,对于HOMO-1、HOMO、LUMO和LUMO+1四个轨道有不同程度的影响。2-Cl-FBP中的氯原子主要影响LUMO和LUMO+1轨道。同时氯原子对LUMO和HOMO-1有部分的贡献。4-Cl-FBP中的氯原子也是主要影响LUMO和LUMO+1轨道,和2-Cl-FBP所不同的是,氯原子参与了HOMO和LUOMO轨道。2'-Cl-FBP中,氯原子主要影响了HOMO和HOMO-1,氯原子参与了HOMO-1、HOMO和LUMO轨道。通过讨论表明氯原子除了通过影响四个轨道本身来影响光谱的吸收,同时氯原子本身也会参与部分轨道的形成,从而影响吸收光谱。
2.3 溶剂效应
前面的讨论都是基于气态这一条件,溶剂对物质的光谱有重要影响,这里我们选取了三种极性不同的溶剂(苯、二氯甲烷和水)来研究不同溶剂对电子吸收光谱的影响。图2显示四种物质在气态和不同溶剂中的吸收峰位置和强度的变化。从图中发现虽然三种溶剂极性差别很大,但是所有溶剂都使这四种分子的Soret带发生红移,由350 nm附近移到370 nm左右。有意思的是,所有溶剂对Q带的峰位置影响并不明显。然而,不同体系Q带的强度对于不同的溶剂的响应区别是很大的。对于FBP来讲,随着溶剂极性的增大,Qy依次减小,直到最后到水溶剂下Qy的吸收峰消失。类似的情形也出现在2'-Cl-FBP体系中,在气态下卟啉环上2'位置引入氯原子后Qy的吸收明显比Qx的吸收强,随着溶剂极性越来越大,2'-Cl-FBP体系Qy的吸收越来越弱。2-Cl-FBP体系表现的不是特别明显,这主要在于引入氯原子后使得Qx吸收峰大大增强,而Qy的吸收几乎消失,因此,2-Cl-FBP体系的Q带对不同溶剂的响应较小。4-Cl-FBP体系和2-Cl-FBP有类似的现象。因此,溶剂对于Soret带的峰位置具有重要影响,而溶剂极性对于Qy带的吸收强度有重要影响。
3 结论
综上,本文利用密度泛函理论计算了FBP和三种氯原子取代卟啉的基态结构和分子轨道性质。采用TDDFT方法计算了气态、苯、二氯甲烷和水不同环境下各个分子结构的电子吸收光谱。通过计算发现,氯原子的引入打破了原有的对称性,从而对Q带的吸收强度影响很大。溶剂会使Soret带发生红移,而Soret带对溶剂极性不敏感。溶剂的极性对Qy带的吸收影响较大。本文从理论上揭示了不同氯取代对卟啉光谱的影响,并进一步讨论了不同极性溶剂的影响,所得结论为进一步设计氯取代卟啉光电材料提供理论基础。
猜你喜欢
分子催化(2022年1期)2022-11-02
能源工程(2021年1期)2021-04-13
中国特种设备安全(2020年11期)2020-06-09
中成药(2018年12期)2018-12-29
柴油机设计与制造(2018年3期)2018-10-13
中国资源综合利用(2017年1期)2018-01-22
中南大学学报(自然科学版)(2016年2期)2017-01-19
山东工业技术(2016年15期)2016-12-01
中国粮油学报(2016年5期)2016-01-23
天然产物研究与开发(2014年7期)2014-04-27
