
法舒地尔调控巨噬细胞极化改善糖尿病小鼠心肌纤维化*
2019-05-28 09:05谢发江蒋松辰高尚远冉茂霞李家富
中国病理生理杂志 2019年5期
谢发江,蒋松辰,高尚远,李 燕,冉茂霞,李家富,冯 健
(西南医科大学附属医院心血管内科,四川 泸州 646000)
糖尿病心肌病(diabetic cardiomyopathy,DCM)系糖尿病(diabetes mellitus,D)患者中独立于大血管及微血管病变的特异性心肌损害,是发达国家糖尿病患者致死的主要原因之一[1]。长期高血糖刺激导致心肌细胞肥大及纤维化,早期可出现心脏舒张功能下降,并逐渐累及收缩功能,最终发展为充血性心力衰竭[2]。
炎症和纤维化是DCM发病的重要机制之一,单核-巨噬细胞渗出及分化是炎症启动及调节的关键环节,巨噬细胞受局部微环境的影响至少分化为2种亚型参与炎症反应,这一过程称之为巨噬细胞极化,其中以经典活化型(M1型)巨噬细胞和选择活化型(M2 型)巨噬细胞为主,M1型巨噬细胞上调诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)及炎症因子的表达,过度极化可导致组织损伤,M2型巨噬细胞上调精氨酸酶1(arginase-1,Arg-1)及抗炎因子的表达,发挥抗炎效应,有利于组织修复[3]。M1/M2比例失调促进DCM发生,调控巨噬细胞极化显示了良好的抗纤维化及对DCM的治疗作用[4-7]。
RhoA蛋白是Ras超家族中一种小分子三磷酸鸟苷(guanosine triphosphate,GTP)结合蛋白,其下游效应分子为Rho激酶(Rho-associated kinases,ROCK),后者属于丝氨酸/苏氨酸蛋白激酶家族成员,存在2种亚型: ROCK1(又称ROKβ、p160 ROCK)和ROCK2(也称ROKα)。RhoA/ROCK信号通路调节细胞增殖、迁移和凋亡等生物学行为,扮演分子开关的角色[8]。研究发现,RhoA/ROCK信号通路介导炎症、氧化应激、钙平衡、胰岛素抵抗、心肌细胞凋亡、心肌纤维化和心脏舒缩功能等参与DCM发病,是具有巨大潜力的治疗靶点[9]。法舒地尔能特异性结合ROCK中ATP依赖的激酶结构域并抑制其活性,是目前临床上唯一批准使用的ROCK抑制剂,因其强大的扩血管作用而被广泛应用于蛛网膜下腔出血和缺血性心脏病等血管痉挛性疾病的治疗中[10-11]。尽管动物实验结果显示法舒地尔能够抗心肌纤维化进而治疗DCM[12-13],但其深入机制是否与通过调控巨噬细胞极化相关尚无相关文献报道。本研究通过链脲佐菌素(streptozotocin,STZ)诱导1型糖尿病小鼠模型并给予法舒地尔干预,观察其对巨噬细胞极化及心肌纤维化的影响。
材 料 和 方 法
1 实验动物、药品与试剂
60只SPF级健康雄性C57BL/6小鼠,6~8周龄,体重(20.6±0.8)g,购自成都达硕实验动物有限公司,合格证编号为SCXK(川)2015-030。实验期间自由饮水,进食普通饲料,室温(23±1)℃,每日12 h光照维持昼夜循环。
链脲佐菌素购自Sigma;盐酸法舒地尔(hydroxyl fasudil,HF)注射液购自天津红日药业股份有限公司(批准文号为国药准字H20040356);抗CD11c和CD206抗体均购自Proteintech Group;抗肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)、白细胞介素6(interleukin-6,IL-6)和IL-10抗体均购自Bioworld Technology;抗p-MYPT1 Thr853、iNOS和Arg-1抗体均购自CST;抗β-actin抗体、山羊抗兔II抗和山羊抗鼠II抗购自Abcam;BCA蛋白浓度测定试剂盒购自南京凯基生物公司。
2 方法
2.1糖尿病小鼠模型的建立、分组及处理 所有动物适应性喂养1周,随机分为正常组(normal saline组,NS组)、正常+法舒地尔组(N+HF组)、糖尿病组(D+NS组)、法舒地尔低剂量组(D+LHF组)、法舒地尔中剂量组(D+MHF组)和法舒地尔高剂量组(D+HHF组),每组各10只。D+NS组及各治疗组小鼠禁食12 h后,腹腔注射1% STZ (80 mg/kg),NS组和N+HF组小鼠腹腔注射等体积0.1 mmol/L柠檬酸钠缓冲液,连续7 d,2周后断尾取血,测得血糖值≥16.7 mmol/L视为造模成功。成模后各治疗组小鼠分别腹腔注射低剂量(10 mg/kg)、中剂量(40 mg/kg)和高剂量(60 mg/kg)法舒地尔,每天1次[14],N+HF组予中剂量(40 mg/kg)法舒地尔,NS组和D+NS组腹腔注射等体积生理盐水,连用8周,复测随机血糖及体重,腹腔注射1%戊巴比妥钠麻醉,开胸取心脏,将其横向对半切开,心尖部组织用4%多聚甲醛固定后制作成厚4 μm石蜡切片,心底部组织冻存于-80 ℃冰箱,用于Western blot检测。
2.2心脏组织病理学观察 分别选取各组石蜡切片,脱蜡至水,行HE及Masson染色,于光镜下观察拍照。采用图像分析软件Image-Pro Plus 6.0进行心肌胶原容积分数(collagen volume fraction,CVF)测定,CVF(%)=胶原面积/视野总面积×100%。
2.3免疫组化观察心脏组织中巨噬细胞极化及炎症因子和抗炎因子水平 将切片脱蜡后浸入0.01 mmol/L柠檬酸盐缓冲液(pH 6.0),微波炉中高火加热至沸腾后断电,间隔5 min后,重复1次,冷却后,PBS洗2次,每次5 min,进行抗原修复,并滴加山羊血清室温封闭20 min。分别加入抗CD11c(1 ∶50)、CD206(1 ∶100)、IL-6(1 ∶100)、TNF-α(1 ∶100)和IL-10(1 ∶50)抗体,4 ℃孵育过夜,滴加 II 抗,37 ℃继续孵育30 min,PBS洗3次,每次5 min,DAB显色。最后用苏木素复染,脱水,中性树胶封片。IL-6、TNF-α和IL-10染色切片于100倍镜下选取6个不同观察区域,并进行400倍镜下拍片,采用Image-Pro Plus 6.0图像分析系统测定所采集6个视野全部图像的积分吸光度(integrated absorbance,IA)和阳性表达面积(stained area,SA),并计算每张图像的平均吸光度(mean absorbance,MA),使用6张图像的平均吸光度再计算平均数,得出每例样本的平均吸光度,再进行分析,按同样的方式测算CD11c和CD206阳性细胞数。
2.4Western blot测定蛋白水平 取各组小鼠心脏冻存组织样本,37 ℃水浴解冻后按质量体积比1 ∶10加入RIPA 裂解液,快速剪碎组织,置碎冰上裂解10 min,收集裂解液,4 ℃、12 000 r/min离心10 min,取上清液,按BCA蛋白定量试剂盒说明测定蛋白浓度。根据所需样本体积,按4:1比例加入5×Loding Buffer,混匀后热循环仪95 ℃、 15 min使蛋白变性。配制8%、10%分离胶和5%浓缩胶,按蛋白量60 μg上样,进行电泳分离,转PVDF膜,经5% BSA液封闭2 h后,加入抗p-MYPT1 Thr853(1∶500)、iNOS(1 ∶200)、Arg-1(1 ∶300)及β-actin (1 ∶5 000)抗体,4 ℃孵育过夜,TBST(pH 7.4)洗膜3次,每次5 min,将膜放入相应 II 抗(1 ∶5 000)中,室温孵育2~3 h,TBST洗膜3次,每次10 min,ECL发光液显色,采用ChemiDoc XRS凝胶扫描成像仪及Image Lab凝胶分析系统进行条带分析。
3 统计学处理
采用SPSS 17.0统计软件进行统计分析。数据以均数±标准差(mean±SD)表示,多组样本均数间比较采用单因素方差分析(one-way ANOVA)检验,方差齐则组间采用SNK-q检验,以P<0.05为差异有统计学意义。
结 果
1 各组小鼠体重及血糖的变化
与NS组相比,D+NS组小鼠成模后出现体重明显下降,血糖水平亦明显升高(P<0.05)。N+HF组较NS组体重与血糖值的差异并无统计学显著性。同样,各治疗组体重与血糖值较D+NS组相比差异也无统计学显著性,说明法舒地尔对小鼠体重及血糖无明显影响,见图1。
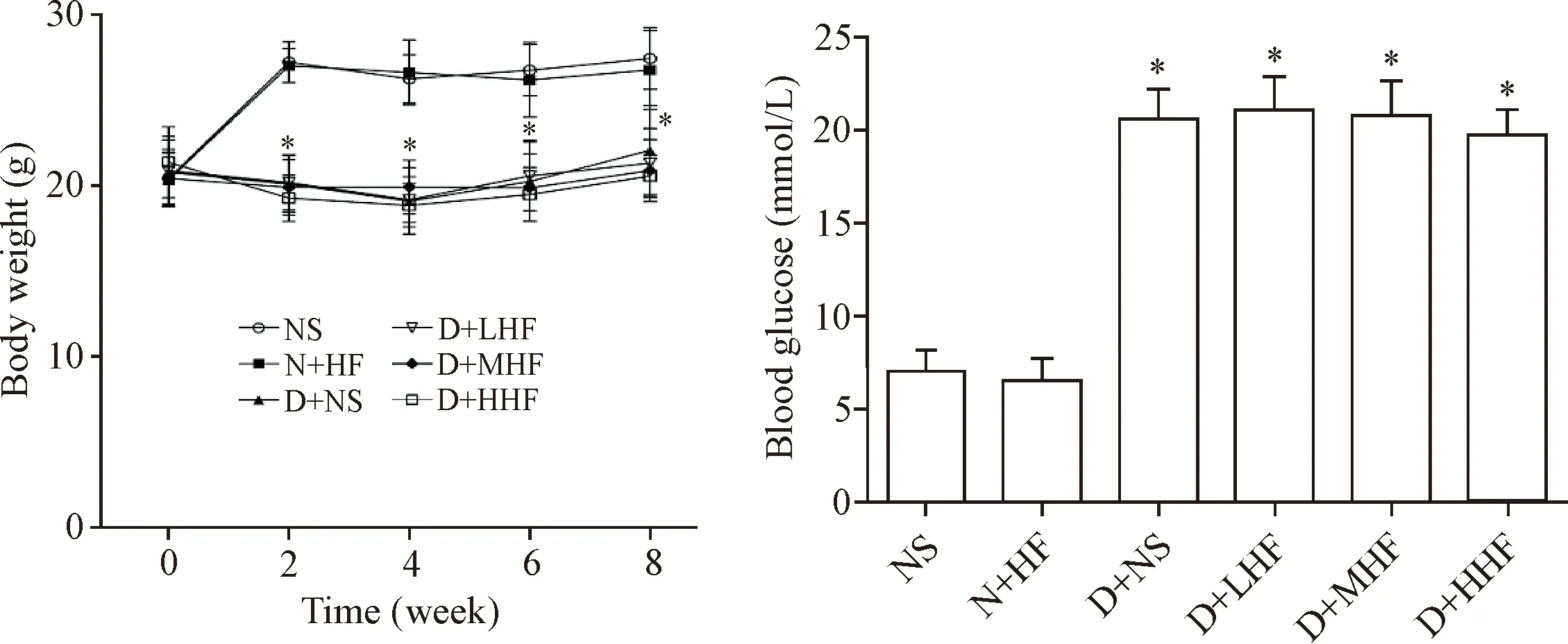
Figure 1.The body weight and blood glucose of the mice in each group were detected. Mean±SD.n=10.*P<0.05vsNS group.
图1 各组小鼠体重及血糖的变化
2 小鼠心脏组织HE染色
HE染色观察可见NS组和N+HF组心肌细胞排列整齐,轮廓清晰,细胞质着色均匀;D+NS组心肌细胞排列紊乱,胞质着色不均,中膜层心肌纤维呈波浪样变性,部分断裂;各治疗组较D+NS组心肌细胞排列较整齐,轮廓大致清晰,细胞质着色较均匀,中膜层心肌纤维波浪样变性及断裂改变明显减轻,见图2。
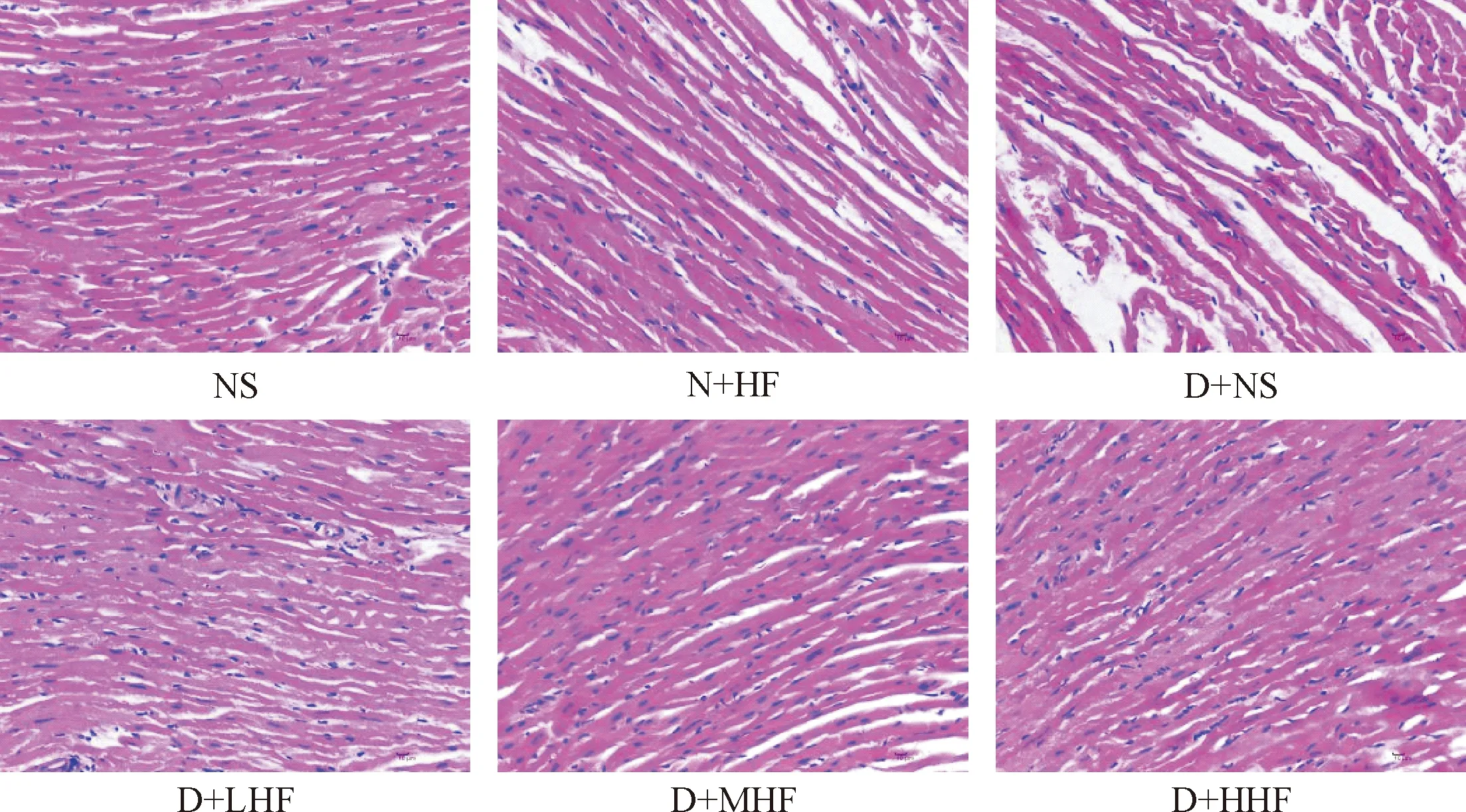
Figure 2.The representative images of cardiac tissues in each group under light microscope (HE staining, ×400).
图2 各组小鼠心脏组织HE染色结果
3 小鼠心脏组织Masson染色及CVF的变化
Masson染色时胶原纤维呈蓝色,心肌细胞胞浆呈红色。NS组和N+HF组心肌间质可见少许胶原纤维分布,CVF的差异无统计学显著性;与NS组相比,D+NS组心肌间质胶原纤维分布明显增加,CVF显著增大(P<0.05);与D+NS组相比,各治疗组心肌间质胶原纤维分布明显较少,CVF下降(P<0.05);与D+LHF组相比,D+MHF组和D+HHF组CVF进一步下降(P<0.05),提示DCM存在明显心肌纤维化,法舒地尔能剂量依赖性地减轻DCM心肌纤维化,见图3。

Figure 3.Masson staining showed the changes of CVF of cardiac tissues in each group (×400). Mean±SD.n=6.*P<0.05vsNS group;#P<0.05vsD+NS group;$P<0.05vsD+LHF group.
图3 Masson染色观察各组小鼠心脏组织的CVF变化
4 免疫组化观察小鼠心脏组织中巨噬细胞极化及炎症因子和抗炎因子的水平
CD11c识别M1型巨噬细胞,CD206识别M2型巨噬细胞。NS组和N+HF组中M1和M2型巨噬细胞数量及IL-6、TNF-α和IL-10的蛋白水平差异无统计学显著性;与NS组相比,D+NS组M1型巨噬细胞数量增加,M2型巨噬细胞数量减少,IL-6和TNF-α的蛋白水平显著增加(P<0.05),IL-10的蛋白水平显著下降(P<0.05);与D+NS组相比,各治疗组的M1型巨噬细胞数量及IL-6和TNF-α的蛋白水平均呈剂量依赖性减少(P<0.05),M2型巨噬细胞数量及IL-10的蛋白水平随着剂量增大而增加(P<0.05),见图4。

Figure 4.Immunohistochemical identification of macrophage phenotype marker proteins and the protein expression of IL-6, TNF-α and IL-10 in different groups (×400). Mean±SD.n=6.*P<0.05vsNS group;#P<0.05vsD+NS group;$P<0.05vsD+LHF group;&P<0.05vsD+MHF group.
图4 各组小鼠心脏组织巨噬细胞极化及炎症因子和抗炎因子蛋白水平的变化
5 小鼠心脏组织中p-MYPT1 Thr853、iNOS和Arg-1的蛋白水平
NS组和N+HF组p-MYPT1 Thr853、iNOS和Arg-1的蛋白水平差异无统计学显著性;与NS组相比,D+NS组p-MYPT1 Thr853和iNOS的蛋白水平升高,Arg-1的蛋白水平降低(P<0.05);与D+NS组相比,D+LHF组p-MYPT1 Thr853、iNOS和Arg-1的蛋白水平差异无统计学显著性;与D+NS组相比,D+MHF组和D+HHF组p-MYPT1 Thr853和iNOS的蛋白水平下降(P<0.05),Arg-1的蛋白水平升高(P<0.05),见图5。
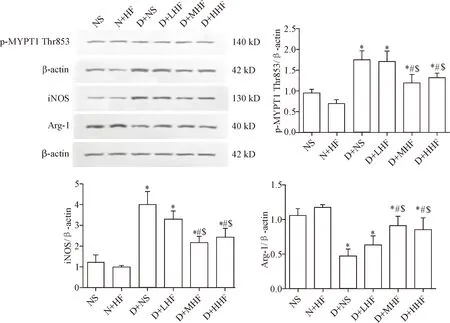
Figure 5.The protein levels of p-MYPT1 Thr853, iNOS and Arg-1 in different groups were determined by Western blot. Mean±SD.n=3.*P<0.05vsNS group;#P<0.05vsD+NS group;$P<0.05vsD+LHF group.
图5 各组小鼠心脏组织中p-MYPT1 Thr853、iNOS和Arg-1的蛋白水平变化
6 相关性分析
通过两变量间相关性分析(Pearson相关分析)发现,小鼠心脏组织中p-MYPT1 Thr853的蛋白水平与iNOS的蛋白水平明显呈正相关(r=0.914,P<0.01),与Arg-1的蛋白水平明显呈负相关(r=-0.887,P<0.01),见图6。
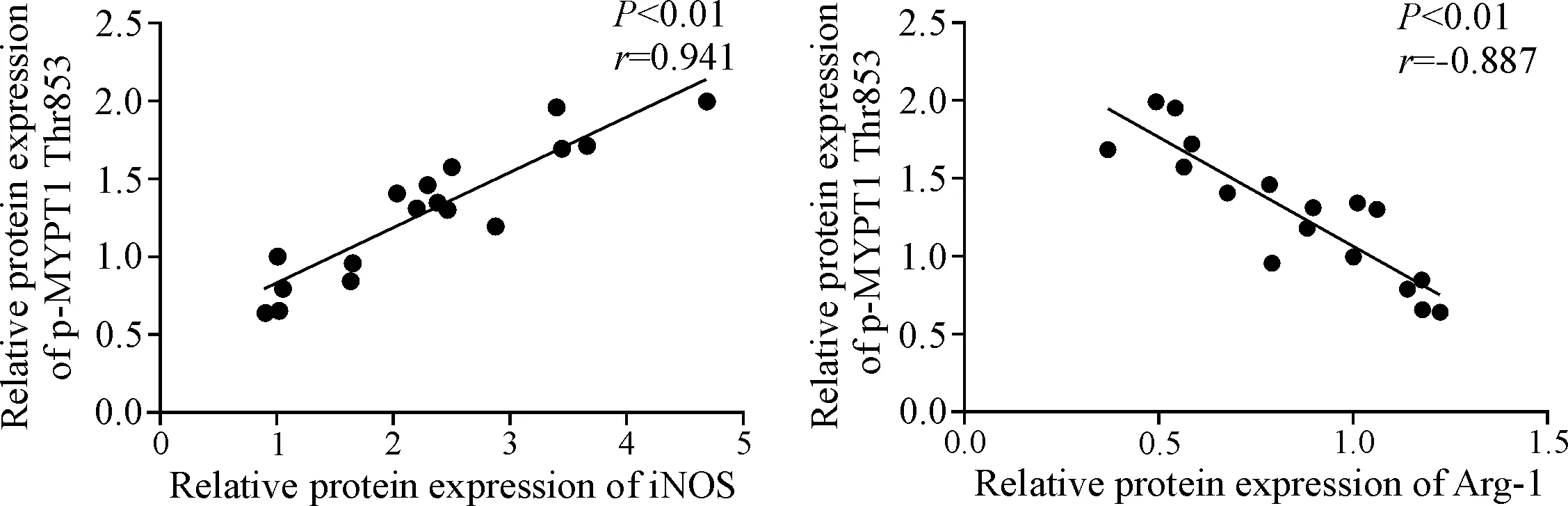
Figure 6.Correlations between p-MYPT1 Thr853 and iNOS or Arg-1 expression.
图6 各组小鼠心脏组织中p-MYPT1 Thr853的蛋白水平分别与iNOS和Arg-1蛋白水平的相关性
讨 论
在病理状况下,单核细胞由循环系统迁移并渗出到组织中分化形成巨噬细胞。未分化M0型巨噬细胞在不同微环境下可诱导分化成不同类型。M1型巨噬细胞主要受脂多糖(lipopolysaccharides,LPS)及干扰素γ(interferon-γ,IFN-γ)诱导,此外促炎因子TNF-α和IL-12等也能产生诱导作用。活化的M1型巨噬细胞分泌大量的活性氮,加强Th1细胞免疫,随着大量炎症介质的释放产生强大的抗菌及细胞毒性作用,具有抗感染及抗肿瘤效应,在急性损伤炎症初期,首先渗出起到清除坏死组织的作用[15]。M2型巨噬细胞受诱导因子不同可分化为M2a、M2b和M2c 3种不同功能的亚型,M2a型由IL-4和IL-13诱导,M2b型由免疫复合物和IL-1诱导,IL-10、转化生长因子-β(transforming growth factor-β,TGF-β)和糖皮质激素诱导形成M2c型。M2a型能抑制INF-γ、IL-1和IL-6等炎症介质的释放,同时能促进TGF-β表达及细胞间基质的沉积,但其吞噬功能较差,因具有修复组织的功能而又被称为“组织修复型巨噬细胞”;M2b和M2c型通过介导慢性炎症限制M1型巨噬细胞的异常持续活化造成的组织破坏,并未直接参与组织修复,而被称为“调节型巨噬细胞”[16]。在炎症的不同阶段M1和M2型巨噬细胞发挥不同的作用,M1/M2型巨噬细胞比例的动态变化决定了炎症的进展及严重程度。Urbina等[5]发现在糖耐量减低小鼠模型的心脏组织中M2型巨噬细胞与正常组相比差异并无统计学显著性,但经骨形成蛋白7(bone morphorgenic protein-7, BMP-7)干预的模型小鼠M2型巨噬细胞数量较对照组明显升高,同时伴有促炎因子TNF-α和IL-6降低及抗炎因子IL-1RA和IL-10的升高,干预组心肌纤维化及心脏功能明显改善。后续研究发现,DCM心脏组织中巨噬细胞以M1型为主,过度活化的M1型巨噬细胞加重心脏损伤,当完全抑制小鼠中巨噬细胞的增殖及分化时,DCM病变并未有效减轻,但通过诱导M2型巨噬细胞极化,抑制M1型巨噬细胞的极化,促炎因子明显下降,抗炎因子升高,DCM的病变明显减轻[4]。越来越多的证据表明,调控M1/M2型巨噬细胞的比例是DCM治疗有效靶点[6-7]。本实验中,我们选择CD11c和iNOS作为M1型巨噬细胞的标志物,CD206和Arg-1作为M2型巨噬细胞的标志物,TNF-α和IL-6为观察的促炎因子,IL-10为观察的抗炎因子。与正常组相比,糖尿病组小鼠中M1型巨噬细胞数量及促炎因子的表达明显增加,M2型巨噬细胞数量明显减少,而通过法舒地尔的干预M1型巨噬细胞极化明显抑制,M2型噬细胞数量增加,炎症水平明显下降,DCM心肌纤维化有效改善,且随着剂量增加抗纤维化作用越明显,提示法舒地尔调控巨噬细胞极化,恢复异常的M1/M2型巨噬细胞的比例,可能是其抗心肌纤维化的机制之一。
大量的研究表明,法舒地尔能有效改善DCM,目前认为其可能与下列机制有关:(1)抑制纤维化关键信号通路JNK 和TGF-β/Smad的表达,减轻间质纤维化[13];(2)提高心肌细胞线粒体中琥珀酸脱氢酶(succinate dehydrogenase,SDH)和单胺氧化酶(monoamine oxidase,MAO)的活性,抑制脂质氧化终产物丙二醛(malondialdehyde,MDA)的生成,提高抗氧化水平,并通过减少线粒体膜通透性转运孔(mitochondrial permeability transition pore,MPTP)开放数量,保护线粒体结构与功能[17];(3)抑制心肌细胞Bax 表达,促进Bcl-2表达,减轻心肌细胞凋亡[12, 18];(4)提高心肌细胞舒张期Ca2+回收相关转运蛋白的表达与活性,促进舒张期胞质Ca2+回收,并且修复受损的肌动蛋白与横桥空间结构,直接改善心肌的舒缩功能[19-20]。法舒地尔能特异性结合ROCK中ATP依赖的激酶结构域并下调其活性,进而抑制RhoA/ROCK信号通路转导,MYPT1是ROCK下游结合底物之一,MYPT1的磷酸化程度能有效反映ROCK的活性[21]。本研究中,糖尿病组小鼠p-MYPT1 Thr853的蛋白水平增加,提示DCM中存在RCOK的激活,经法舒地尔干预后,p-MYPT1 Thr853的蛋白水平下降,RCOK的活性受到抑制,同时我们发现MYPT1 Thr853磷酸化水平与iNOS的蛋白水平呈正相关,反之与Arg-1的蛋白水平呈负相关,说明ROCK活性与巨噬细胞极化存在相关性。RhoA/ROCK信号通路过度激活是DCM发病的关键机制之一[22]。Cheng等[23]发现高糖状态下培养的巨噬细胞中RhoA/ROCK信号通路异常激活并调控巨噬细胞的黏附和分泌等多种生物学功能。由此我们推测,DCM发病中巨噬细胞的极化可能受RhoA/ROCK信号通路的调控,其机制有待进一步体外研究。
综上所述,法舒地尔可能通过抑制M1型巨噬细胞极化,诱导M2型噬细胞极化,下调心脏炎症水平,进而减轻DCM心肌纤维化,有望成为DCM治疗的一种新策略。同时,本实验对DCM发病中RhoA/ROCK信号通路与巨噬细胞极化的关系做了初步探究,巨噬细胞极化是否受RhoA/ROCK信号通路调控有待进一步研究。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
传染病信息(2022年3期)2022-07-15
现代财经-天津财经大学学报(2022年5期)2022-06-01
世界科学技术-中医药现代化(2022年2期)2022-05-25
航天电子对抗(2022年2期)2022-05-24
北京航空航天大学学报(2021年9期)2021-11-02
昆明医科大学学报(2021年2期)2021-03-29
中国生殖健康(2020年7期)2020-12-10
家庭医药(2019年8期)2019-08-27
航天电子对抗(2019年4期)2019-06-02
