
基于XGBoost的RNA修饰位点的识别
2019-05-25 11:26:18吕成伟樊永显
桂林电子科技大学学报 2019年6期
吕成伟, 樊永显
(桂林电子科技大学 计算机与信息安全学院,广西 桂林 541004)
自经过修饰的RNA核糖核酸第一次发现以来,被人类已知的RNA修饰类型已经达到了约150种[1]。研究表明,RNA修饰是基因调控的关键组成部分[2],其参与了转录后的各种生物过程,如蛋白质翻译和定位、mRNA剪接等,并发挥着重要作用[3]。但是,RNA修饰在其他方面的功能对人们来说仍然是未知的。因此,预测RNA修饰位点对于理解它们的分子机制和功能起着至关重要的作用。
新一代测序技术的出现为在全基因组范围内研究RNA修饰提供了契机。如N1-甲基甘氨酸(m1A)、N6-甲基甘氨酸(m6A)和5-甲基胞嘧啶(m5C)图谱可用于人类转录组。虽然这些基于生物实验技术的高通量测序方法在理解生物功能和RNA修饰方面起到了积极推动的作用,但这些方法有很大局限性,其实验成本高、耗时长。为了解决该问题,一些用于识别RNA修饰位点的基于高分辨率实验数据的计算方法被提出。针对m6A修饰位点的识别问题,Chen等[4]提出了基于序列的iRNAMethy方法,使用了伪二核苷酸组分(pseudo dinucleotide composition,简称PseDNC)编码方式,在特征提取方式上取得了一些突破。Chen等[5]在编码方式上进行了创新,提出了m6Apred方法,在原有序列信息的基础上计算出核苷酸的频率信息,且加入了其化学分类特征,进一步提高了m6A修饰位点的识别准确率。Zhang等[6]提出了一种新的提取特征的方法m6A-HPCS,该方法的思想与Chen等[5]提出的m6Apred基本一致,从23种核苷酸物理化学性质中出寻找一个最优子集,结合自协方差和互协方差变换提取序列特征。
上述几种方法为基于序列的RNA修饰位点的预测开辟了道路,并取得了一定的成功。但这几种方法对m6A和m5C修饰位点的识别准确率不够理想,仍有较大的提升空间。针对上述问题,在采用PseKNC的编码方式对样本序列进行编码的基础上,添加了位置特异性单核苷酸及二核苷酸偏好特征,并基于XGBoost集成算法构建了预测模型。实验证明,该模型的鲁棒性好,且针对上述提出的3种RNA修饰位点的预测都取得了较高的准确率。
1 材料与方法
为了开发一种新的预测方法,文献[7-11]遵循了Chou[12]提出的一些原则,并明确以下4个步骤:
1)构造一个高质量的基准数据集用来训练和测试预测模型;
2)对生物序列进行编码,使其能够被预测模型识别;
3)选择或者自主研发一种鲁棒的算法来建立预测模型;
4)进行交叉验证,并客观地评价预测模型的好坏。
1.1 基准数据集
本研究所使用的数据集包括m1A、m6A、m5C 3种RNA序列[13],这3种RNA序列分别包含6 366、1 130和120个正样本。为了平衡正负样本,从对应的负样本中随机选取了6 366、1 130和120个样本分别作为m1A、m6A和m5C的负样本。
1.2 构建RNA序列样本
生物信息学面临的一个极具挑战的问题是:如何对生物序列进行有效编码,使得其编码后的序列尽可能包含序列模式特征,并能被现有的机器学习模型直接识别。几乎所有的机器学习算法都是为了处理向量而开发的,并不能直接识别序列样本。常用的独热编码虽然能解决不能被机器学习模型识别的问题,但这种编码方式会丢失许多重要的序列模式特征,从而导致最终的预测模型预测能力低下。为了解决蛋白质序列的问题,研究人员开发了伪氨基酸组成物(pseudo amino acid composition,简称PseAAC)[14-18]。自PseAAC引入以来,它的概念几乎渗透到计算蛋白质组学的每个领域。在PseAAC概念的启发和巨大成功的鼓舞下,PseKNC[28]被提出来,并应用到基因组分析的各个领域。
假设RNA序列由L个核苷酸残基组成,
S={R1,R2,…,RL},
(1)
其中R1、R2分别为RNA序列的第1、2个核苷酸残基。
本研究用PseKNC对RNA序列进行编码,最终得到具有4k个分量的向量,即
(2)
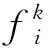
对于式(2),当k=2时,表示RNA序列由二核苷酸配置而成,于是有
D=[f(AA)f(AC)f(AG)…f(UU)]T=
(3)
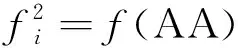
D=[f(AAA)f(AAC)f(AAG)f(UUU)]Τ=
(4)
1.3 XGBoost
XGBoost是一种基于GBDT(gradient boosting decision tree,简称GBDT)梯度下降框架的集成学习算法。GBDT是将梯度下降和决策树相结合,基于前一个分类器残差减少的方向上,构造新的分类器,通过多次迭代构造一组弱分类器,对弱分类器输出结果进行加权累加,累加结果作为强分类器输出[21]。XGBoost与GBDT相比,其优点在于改变了GBDT基于Boosting串行序列化求解问题的方式,利用CPU多线程分布式并行计算,并通过对残差进行泰勒二次展开进行求解,从而打破了现有库的计算速度和精度,使得数据处理和运算的速度得到了提升。
造成XGBoost模型精度高、运行速度快的另一个因素是选用分类回归树(classification and regression tree,简称CART)作为决策树。由于CART树的叶子节点对应的值是一个实际的分数,而非一个确定的类别,这使得优化算法的实现变得更加高效。将XGBoost模型表示为如下数学形式:
(5)
其中:F为所有可能的CART树的集合;f为一棵具体的CART树;K为树的棵数。该XGBoost模型由k棵CART树组成。
1.4 性能评估
通过如下步骤判断一个模型的优劣:
1)采取交叉验证的方法测试模型。如文献[22]采用交叉验证的方法对模型进行测试,得到了广泛的认可和使用。交叉验证对于本实验的模型测试也同样不失为一种好的方法。为了减少随意性,并使得实验结果更准确,采用10次十折交叉验证。
2)采用Chou[12]在研究信号肽预测中使用的4个度量参数评价模型。根据文献[8,23-24]中的定义,敏感性N、特异性P、准确率A和马修斯相关系数M分别表示为:
(6)
其中:NTP为含有修饰位点的样本被正确预测为含有该修饰位点数;NTN为不含有修饰位点的样本被正确预测为不含有该修饰位点数;NFP为不含有修饰位点的样本被错误预测为含有该修饰位点数;NFN为含有修饰位点的样本被错误预测为不含有该修饰位点数。
1.5 位置特异性核苷酸偏好特征
位置特异性偏好思想在生物信息学得到了广泛应用,在功能位点的识别及启动子位点的识别方面都取得了非常不错的效果[25-27]。其原理是统计生物序列中某些关键位置或某种核苷酸出现的概率[26],将得到的概率值作为位点识别的特征。受此启发,将位置特异性单核苷酸和双核苷酸偏好特征特征应用到RNA修饰位点中。
1.5.1 位置特异性单核苷酸偏好特征
由式(1)可知,每个样本由L个核苷酸组成,对于一个基准数据集的所有样本,可分别计算出第j(j=1,2,…,L)个位置上4种核苷酸出现的概率,并用一个长度为4的向量表示:
(7)
其中,MA,j、MC,j、MG,j和MU,j分别为A、C、G和U这4种核苷酸在第j个位置出现的概率。
将j从1取值到L得到的位置特异性向量Mj组合在一起,构成一个4×L维的位置特异性单核苷酸偏好矩阵M:
(8)
1.5.2 位置特异性双核苷酸偏好特征
取2个相邻的核苷酸为一个单元,则式(1)样本序列可表示为
S′={N1,N2,…,NL-1},
(9)
其中,Nj={Rj,Rj+1},j=1,2,…,L-1表示第j个位置双核苷酸的类型,而双核苷酸的种类共有16种,即Nj∈{AA,AC,AG,AU,CA,…,UU}。
与M的计算过程类似,可计算出一个维度为16×(L-1)的位置特异性双核苷酸偏好矩阵:
(10)
1.6 特征选择
虽然用PseKNC对RNA序列进行编码是一个不错的选择,但这种编码方式有一个缺点,即数据样本维度会出现爆炸式的增长,从而会出现以下问题:1)容易造成过拟合导致模型的泛化能力偏低;2)信息冗余和噪声会导致模型准确率低下,达不到预期效果;3)高维度样本使得计算机的运算负荷加重,从而使模型的运行时间大大增加。
为了解决上述问题,对数据样本进行特征选择,进而降低样本维度。本研究采用F-score[28]特征选择方法,定义如下:
(11)
2 结果
2.1 PseKNC编码方式中最优k值的确定
对m1A、m6A、m5C这3种RNA序列采用PseKNC的编码方式进行编码,但k取不同的值时,结果也不同,为了找到最优的k值,对k=2,3,4,5,6,7时分别进行编码,其十折交叉验证的结果如图1所示。
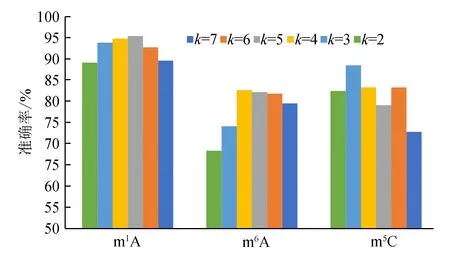
图1 PseKNC编码中不同的k值对m1A, m6A 和 m5C修饰位点的识别准确率的影响
从图1可看出,k取不同值时,m1A、m6A、m5C这3种RNA修饰位点的识别准确率也随之波动,其中m1A对于不同的k值波动较小,而k值的不同对m6A和m5C的影响较大。当k=5时,m1A修饰位点的识别准确率最高;k=4时,m6A修饰位点的识别准确率最高;k=3时m5C修饰位点的识别准确率最高。在进行特征选择之前,m1A、m6A、m5C的PseKNC编码中k的最优值分别为5、4、3。
为了进一步提高识别准确率,采用F-score特征选择方法得到k最终的最优解。
在m1A、m6A、m5C取不同k值(k=2,3,4,5,6,7)编码后的序列基础上,融合位置特异性单核苷酸偏好特征和位置特异性双核苷酸偏好特征,再对其进行特征选择。十折交叉验证的结果如表1所示。
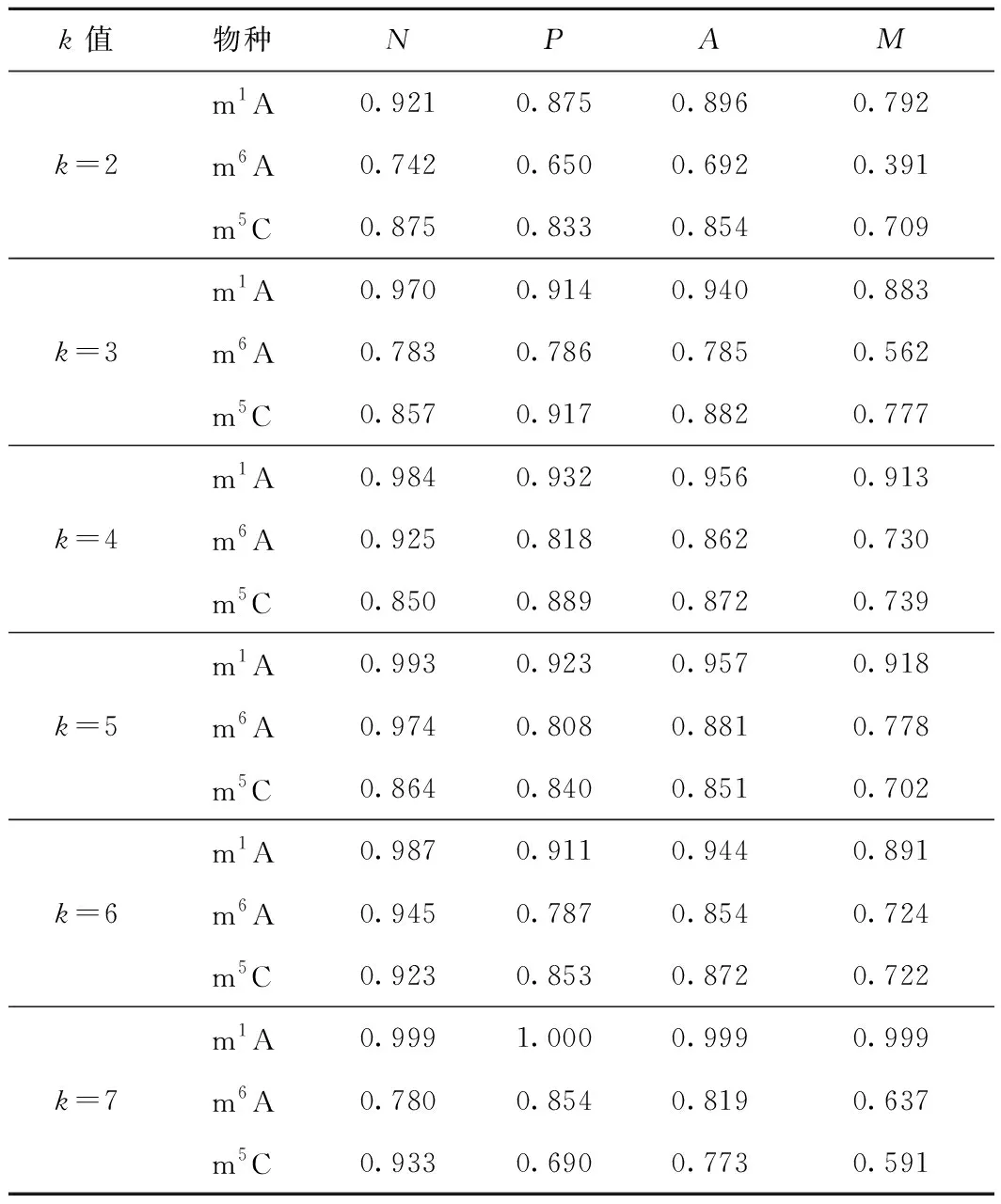
表1 特征选择后,不同的k值对m1A、m6A、m5C修饰位点的识别结果
从表1可看出,融合了位置特异性单核苷酸偏好特征和位置特异性双核苷酸偏好特征并经过特征选择后,m1A、m6A、m5C这3种RNA修饰位点的识别准确率均有较大提升。其中:m1A在k=7时,识别准确率达到最高,为99.9%;m6A在k=5时,识别准确率达到最高,为88.1%;m5C在k=3时,识别准确率达到最高,为88.2%。而在此之前,m1A、m6A、m5C这3种RNA修饰位点的识别准确率最高时其PseKNC编码对应的k值分别为5、4、3,显然,除了m5C的PseKNC编码中最优k值未发生改变,其他2个均发生了改变。
2.2 基于网格搜索的XGBoost模型参数寻优
在确定了PseKNC编码的最优k值后,继续对XGBoost预测模型的参数进行寻优。要想完全发挥XGBoost的强大性能,对其进行调参是必不可少的一项工作。这里选用网格搜索的方法对其进行调参。网格搜索的原理是:在所有候选的参数中,通过循环遍历,尝试每种可能性,交叉验证后,表现最好的参数组合就是最终结果。该方法的优点是结果准确,但缺点是当参数数量过多时,参数寻优的计算过程非常耗时。XGBoost中的参数主要分为通用参数、学习任务参数和命令行参数3大类。其中通用参数有20多个,学习任务参数有4个,命令行参数有十多个,若对这些参数进行网格搜索寻优,将会非常耗时,使调参的工作面临巨大挑战。
为了应对这一挑战,针对性地选取一些核心参数,将对模型性能影响不大的参数剔除。据此,在通用参数中选取booster、learning_rate、max_depth、min_child_weight、subsample、colsample_bytree、gamma、lambda和alpha这8个参数,在学习任务参数中选取objective参数,在命令行参数中选取num_round参数。其中:max_depth和min_child_weight对决策树的构建起约束作用;subsample和colsample_bytree均是关于采样的参数;lambda和alpha均是正则项。为了进一步减少运算量,采用组合分批网格搜索的策略,即将具有相同作用的参数组合在一起,对组合进行网格搜索,将最优组合与其他参数组合在一起,再进行网格搜索。调参后的XGBoost模型在jackknife测试下的识别结果如表2所示。从表2可看出,m6A修饰位点的识别准确率从88.1%提升到了92.6%,m5C修饰位点的识别准确率从88.2%提升到了89.6%,虽然m1A修饰位点的识别准确率并未得到提升,但在此之前已经达到了99.9%这样一个非常理想的水平。
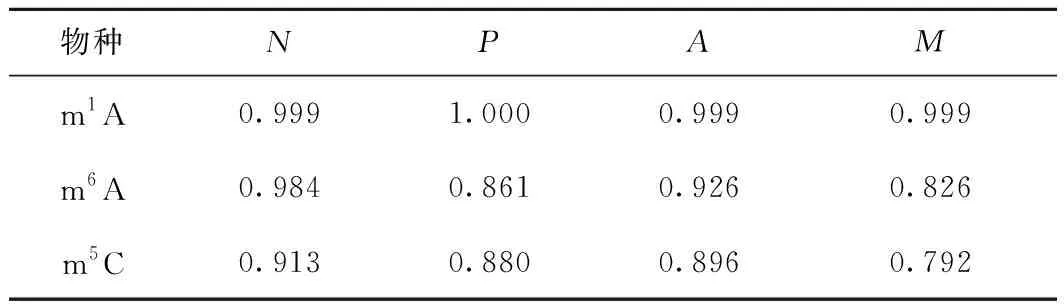
表2 参数调整后的XGBoost模型的识别结果
ROC(receiver operating characteristic)曲线是一个能直观展现模型性能的另一个重要指标[29]。ROC曲线下侧包含的面积(AUC)越大,模型的性能越好[30]。图2为XGBoost预测模型分别对m1A、m6A、m5C这3种RNA修饰位点进行识别后生成的ROC曲线。从图2可看出,m1A、m6A、m5C所对应的AUC值分别为0.998 6、0.931 2和0.955 8,表明XGBoost预测模型的鲁棒性非常好。
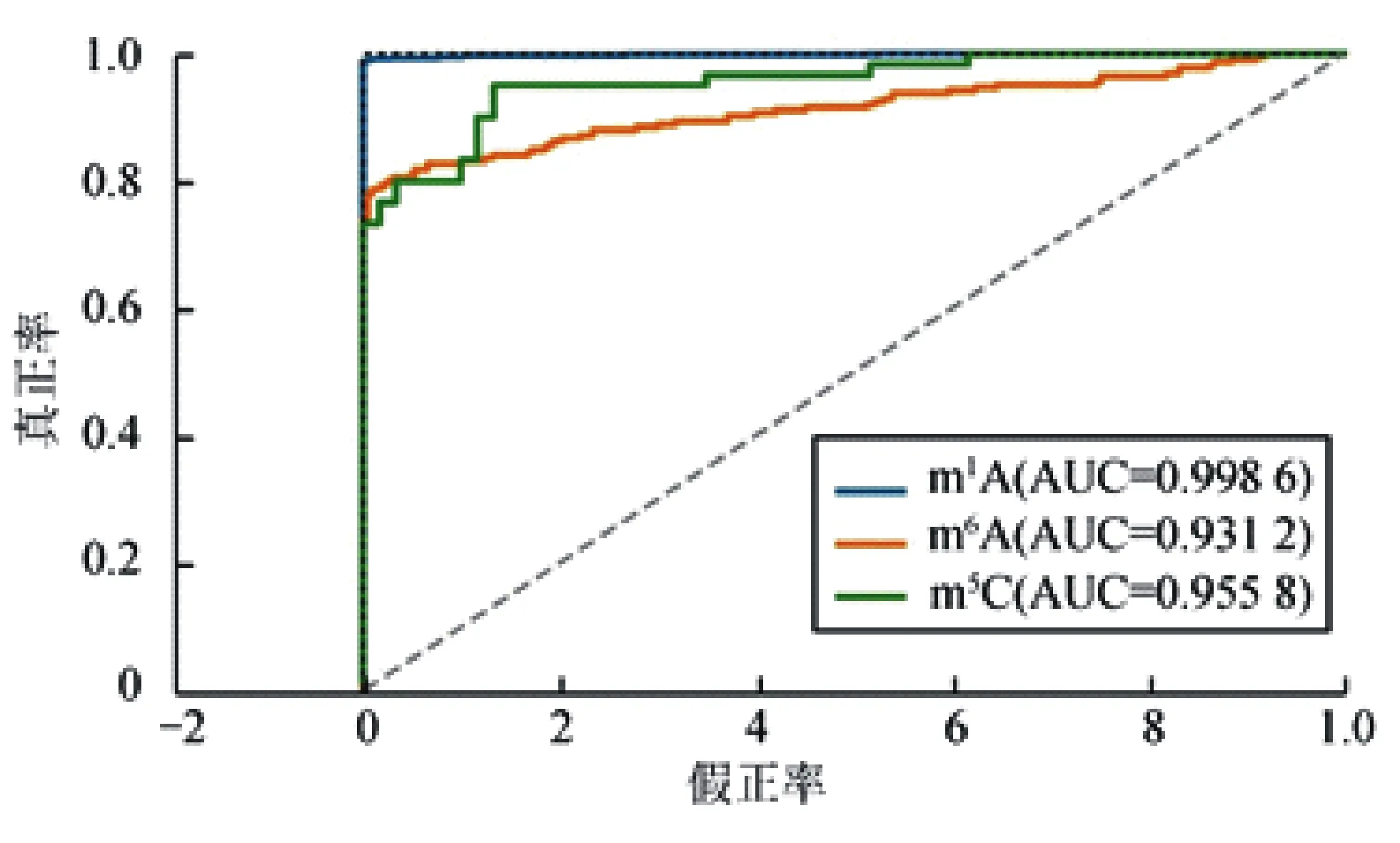
图2 XGBoost模型在m1A,m6A和m5C修饰位点上的识别性能
2.3 不同方法的识别结果对比
将XGBoost预测模型的识别结果与文献[13]使用的SVM预测模型的识别结果进行对比,2个模型经过jackknife测试后的结果如表3所示。从表3可看出,XGBoost预测模型和SVM预测模型在m1A修饰位点的识别上均取得了较好的结果,准确率分别达到了99.9%、99.1%;在m6A修饰位点的识别上,XGBoost预测模型的准确率为92.6%,SVM预测模型的准确率为90.4%,提升了2.2%;在m5C修饰位点的识别上,XGBoost预测模型的准确率达到了89.6%,远高于SVM预测模型的77.5%,提升了12.1%。
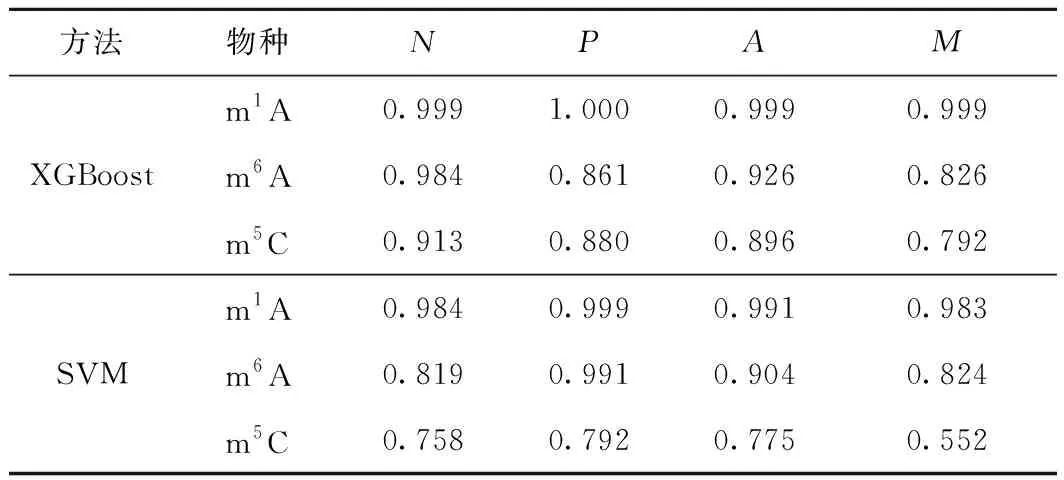
表3 XGBoost与SVM的识别结果比较
3 结束语
为了更快速、准确地识别RNA序列中的修饰位点m1A、m6A、m5C,提出了一种融合位置特异性单核苷酸及双核苷酸偏好特征的PseKNC编码方式,并构建了一个基于XGBoost的RNA修饰位点的预测模型。与现有的SVM预测模型相比,其准确率和马修斯相关系数均取得了明显提升,其中,对于在现有SVM预测模型上识别效果相对较差的m5C修饰位点,在XGBoost预测模型上取得了较大的突破,识别准确率从77.5%提高到了89.6%,马修斯相关系数从0.552提高到了0.792,此外,敏感性和特异性也分别从0.758和0.792提高到了0.913和0.880。XGBoost预测模型的提出为RNA修饰位点的识别提供了高效、可靠的方法。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
肝博士(2022年3期)2022-06-30 02:48:28
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
Journal of Sport and Health Science(2019年6期)2019-11-26 07:30:53
生物学通报(2019年3期)2019-02-17 18:03:58
中国医疗保险(2017年5期)2017-05-17 08:26:39
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
中国康复理论与实践(2015年10期)2015-12-24 05:42:46
现代电生理学杂志(2015年1期)2015-07-18 11:02:16
