
A density functional study on some cyclic N10 isomers Lemi Türker
2019-05-24 01:36MiddleEastTechnicalUniversityDepartmentofChemistryniversitelerEskisehirYoluNo06800ankayaAnkaraTurkey
Defence Technology 2019年2期
Middle East Technical University,Department of ChemistryÜniversiteler,Eskis¸ehir Yolu No:1,06800,Çankaya/Ankara,Turkey
Keyw ords:Nitrogen Polynitrogen Density functional theory Isomers N10
A B S T R A C TPolynitrogen compounds,comprising only nitrogen atoms,are rare.They are considered as promising candidates of clean(green)high energy density materials because of their high energy content and their sole decomposition product is N2.Extending the previous w ork w hich w as on cyclic N8 isomers,in this study some cyclic N10 isomers having 1-4 cycles are considered w ithin the limitations of density functional theory at the levels of B3LYP/6-311++G(d,p)and B3LYP/cc-PVTZ.Some of the structures,including the monocyclic one,decompose by eliminating certain number of N2 units while some remain intact.All the stable isomers(1-3,6-8)investigated presently are highly endothermic that they are candidates for clean high energy materials.Certain quantum chemical properties,IRand UV-VISspectra as w ell as the speci f i c impulse values for the stable structures are reported.
1.Introduction
Although nitrogen is one of the most abundant elements in nature and it forms the highly stable N2molecule,polynitrogen compounds(PNC)w hich comprise only nitrogen atoms are rare and no molecular crystal made of these compounds has been prepared yet[1].Polynitrogen compounds have various allotropic modi f i cations of nitrogen such as N2,N3,N4,etc.Since,they produce only N2gas and have high energy content[2-5]they are considered as promising candidates of clean(green)high energy density materials(HEDM).
Since the early 1990s,potential candidates of polynitrogen compounds have been predicted by the theoreticians and great efforts have been spent in order to synthesize any of them[6-17].Generally,it is believed that employment of polynitrogen compounds w ill have high energetic ef f i ciency to compete w ith liquid propellants[2,18].Based on theoretical calculations and still scarce experimental data,the polynitrogen compounds should have high enthalpy of formation(2-5 kcal/g)and also suf f i ciently high density in the condensed phase(2-4 g/cm3)[18].Additionally,it has been theoretically estimated that the polynitrogen compounds can provide a speci f i c impulse of 350-500 s w ith material density ranging in betw een 2.0-3.9 g/cm3[2].
It is expected in general that polynitrogen molecules should release large amounts of energy w hile decomposing into the very stable N2molecules[19-22].So far HEDMs based on nitrogen atoms have been the subject of several theoretical articles.However,only one solid-state material containing a N5+cation[8,20,23]and a gas phase N5-anion w ere reported experimentally in addition to N3radical and the w ell-know n N3-anion[23].Other species,such as N4,w ere only as short-lived transients structures[22,23].Various topical review s exist portraying the preparative dif f i culty of all-nitrogen compounds[22-25].Recently,tw o breakthroughs in the f i eld has been reported:the synthesis of the N5+cation in a salt[7]and of the cyclo-N5-anion in the gas phase[26].According to theoretical calculations,the cation is V-shaped w hereas the anion is cyclic[24].
Hirshberg et al.,employing PW-DFT w ith the PBE-D[17,27]functional,reported the relative thermodynamic stability,the enthalpies of the N8solid and cg-N form(cubic gauche)at pressures up to 50 GPa[1].Their w ork have revealed the possible existence of such molecular solids,consisting of N8molecules although it is metastable even at ambient pressure[27].DFTtreatment of various cyclic N8structures w ere reported[17].
Mikhailov and Chachkov studied the possibility of the existence of molecular nitrogen allotropes N4,N6,N8,and N10w ith the use of the QCISD and G3 quantum-chemical methods[27].They reported that only N4and N6w ere found to be stable presenting no information particularly about N10.
Previously some cyclic N8isomers have been reported as highly endothermic but stable structures based on the results of DFTcalculations(B3LYP/6-311++G(d,p)and B3LYP/cc-PVTZ)[17].Of those N8isomers,various 4-membered ring containing ones are surprisingly found to be stable.As an extension of that w ork and complete the gap present in the w ork of Mikhailov and Chachkov[27],in the present study,various cyclic N10structures have been considered w ithin the constraints of density functional theory.
2.Method of calculation
Geometry optimizations of all the structures leading to energy minima w ere initially achieved by using MM2 method followed by semi-empirical PM3 self-consistent f i elds molecular orbital(SCF MO)method[28,29]at the restricted level[30,31].Subsequent optimizations w ere achieved at Hartree-Fock level using various basis sets hierarchically.Then,geometry optimizations w ere managed w ithin the framew ork of density functional theory[32,33]and f i nally at the level of B3LYP/6-311++G(d,p)and B3LYP/cc-PVTZ(restricted closed-shell)[30].The exchange term of B3LYP consists of hybrid Hartree-Fock and local spin density(LSD)exchange functions w ith Becke's gradient correlation to LSD exchange[33,34].Note that the correlation term of B3LYPconsists of the Vosko,Wilk,Nusair(VWN3)local correlation functional[35]and Lee,Yang,Parr(LYP)correlation correction functional[36].The vibrational analyses w ere also done.The total electronic energies are corrected for the zero point vibrational energy(ZPE).The stationary points to energy minima w ere proved in all the cases by calculation of the second derivatives of energy w ith respect to the atom coordinates.The normal mode analysis for each structure yielded no imaginary frequencies for the 3N-6 vibrational degrees of freedom,w here N is the number of atoms in the system.This indicates that the structure of each molecule correspondsto at least a local minimum on the potential energy surface.All these calculations w ere done by using the Spartan 06 package program[37].
3.Results and discussion
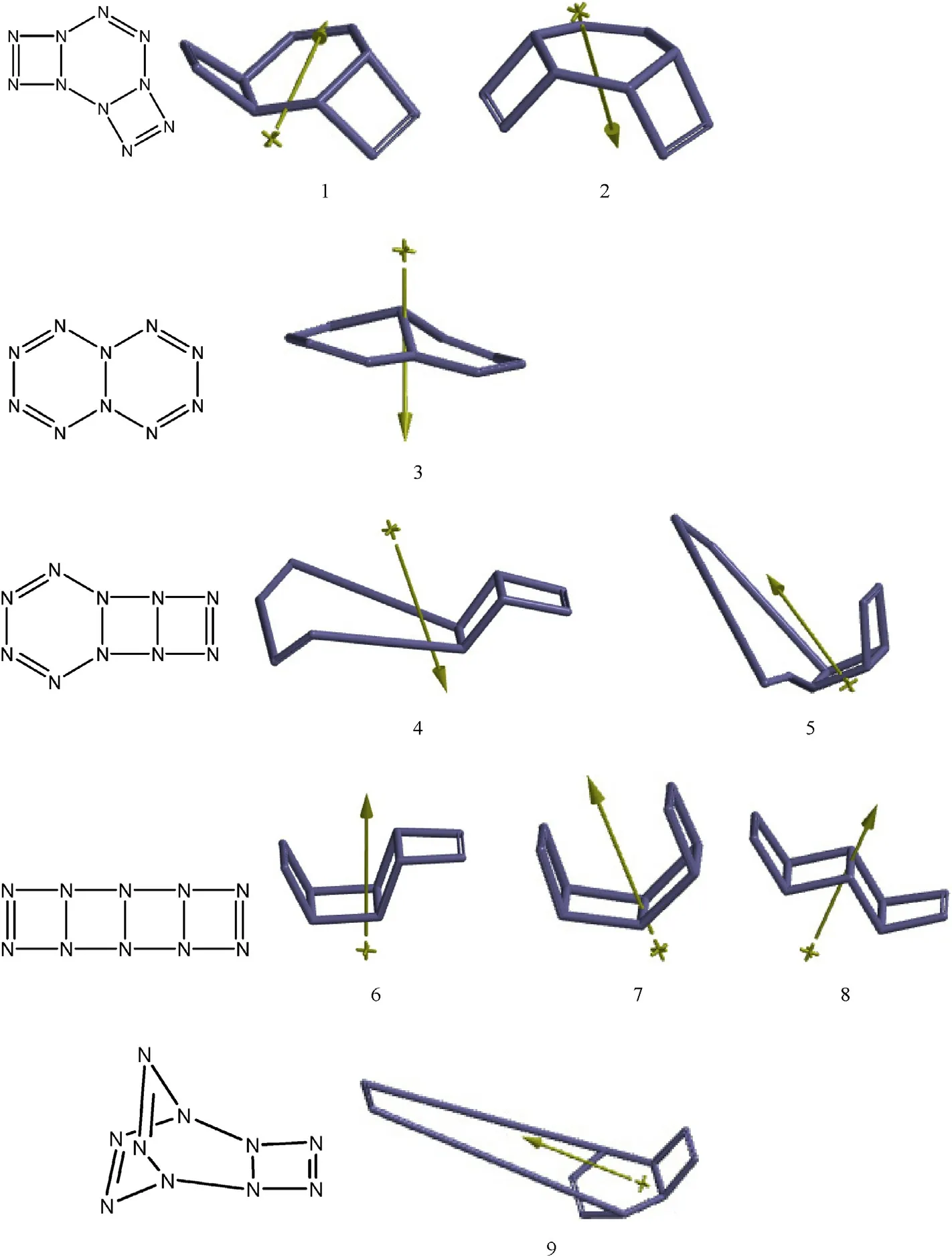
Fig.1.Optimized N10 structures considered(B3LYP/6-311++G(d,p)).
In the present study,the basis sets used are 6-311++G(d,p)and cc-PVTZ.The former one includes diffuse and polarization functions being used for all atoms.Diffuse functions are effective for systems having larger electron density distributions and describing interactions at long distances[38].The second basis set is correlation consistent,designed to describe the correlation of core as w ell as the valence electrons[38].These basis sets as w ell as the functional used(B3LYP)are very popular for the treatment of organic molecules.
3.1.Structures
All the presently considered initial structures are octet stabilized and neutral.The choice of the structures presently considered has been motivated by the valence abilities of nitrogen atom(capable of bonding to one,tw o,or three neighboring atoms through three chemical bonds).Moreover,only cyclic structures having 1-4 cycles have been investigated.Monocyclic N10structure decomposed liberating f i ve N2units but the resultant system failed to converge.Three-cyclic structures 4 and 5 are geometrical isomers of each other and contain 4-membered rings w hich are also unstable and decomposed by eliminating tw o N2units.The remaining fragment in each case is N6structure shaped as an“open book”.Note that this shape of N6w as reported as the stable form[27].Also note that both of the basis sets presently used yield parallel results for stable and unstable structures.Another three-cyclic structure,9 also decomposed expelling a N2unit.Fig.1 shows the optimized structures(including the decomposed ones)and their dipole moment vectors.
3.2.Bond lengths
Fig.2 show s the bond lengths of the isomers presently found to be stable.Note that N-N bond lengths are reported as 1.449Å(hydrazine),1.419Å(1,2-dimethylhydrazine),w hereas N=N bond lengths are 1.254Å(CH3NNCH3),1.219Å(CH3NNCF3)and 1.236Å(CF3NNCF3)[39].
In 1 both of the 4-membered rings have bond lengths identical for the respective bonds.Whereas in its geometrical isomer 2,4-membered rings have different bond lengths,even in the same ring.In structure-6 bond lengths have the same symmetrical occurrence.As for structure-3,the bonds linked to a fusion point have equal lengths,but different than the lengthsof bondsattached to the other fusion point.This result at f i rst glance could be attributed to lone-pair of nitrogen at the fusion point undergoing conjugation,hence causing appearance of some double bond character.How ever,the adjacent bonds next to the assumed double bonds are short(1.24Å)but longer than the other N=N bonds w hich are 1.20Å.Therefore,the bond lengths in these structures should be highly affected by theσ-electron density(see electrostatic charges below)but little by the conjugative effects involving π-electrons.
On the other hand,the N=N bond lengths in all the structures,except structure-3 are in betw een 1.25-1.26Å,w hereas in 3 varies betw een 1.20-1.24Å.The 6-membered rings of structure-3 are more f l exible than 4-membered rings and some conjugative effects betw een theπ-bonds lengthen N=N bonds in 3.Indeed the single bond betw een them is somew hat shorter(1.48Å)than the single bond linked to fusion point(1.52Å).Note that the double bonds in the other structures are located in the 4-membered rings.In structures 6-8,the bond betw een the fusion points vary in the range of 1.40-1.57Å.
So he went to the girl and said, My child, if I do not cut off both thine hands, the devil will carry me away, and in my terror I have promised to do it
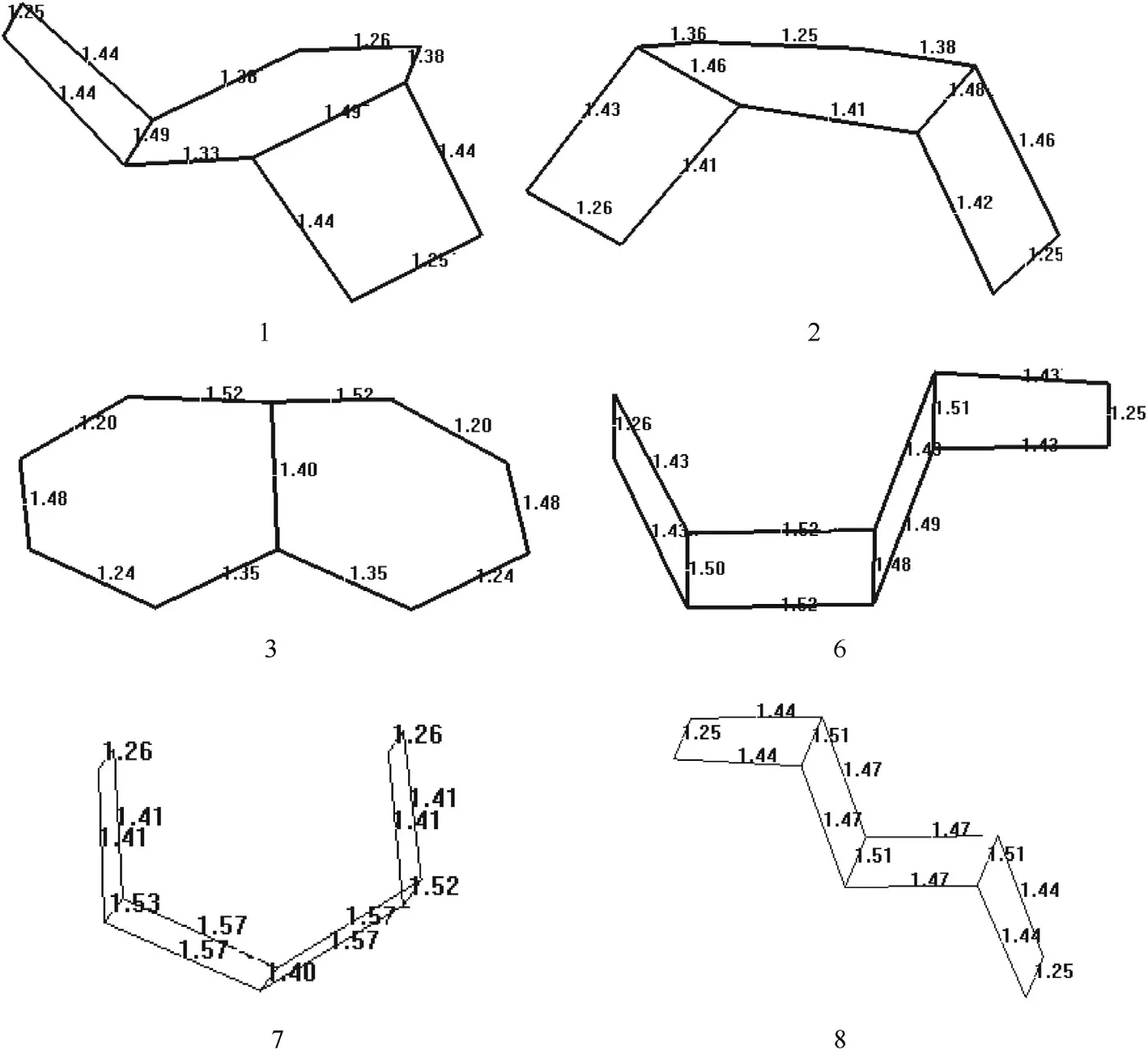
Fig.2.Bond lengths(Å)of the stable isomers considered(B3LYP/6-311++G(d,p).
3.3.Electrostatic charges
Fig.3 displays the electrostatic charge(ESP[37])distribution of the systems of interest.The ESP charges are obtained by the program based on a numerical method that generates charges that reproduce the electrostatic potential f i eld from the entire w avefunction[37].The fusion points in 1 are all positively charged.Whereas in 2(geometrical isomer of 1)the nitrogens at the fusion points betw een the 4-membered ring and 6-memebered ring are oppositely charged.The fusion points in 3 also have opposite charges although their magnitudes are not comparable.In the case of decomposed structures(4,5 and 9)the expelled N2units have only minute charges on the nitrogens w hich could be assumed as molecular nitrogen.
Table 1 show s some properties of the stable isomers presently obtained.Although the stable isomers are structurally different,their areas,volumes and polarizability values are quite comparable w ith each other.The dipole moment values exhibit a scatter,structure-6 and structure-2 having the lowest and highest values,respectively.

Fig.3.Electrostatic charges(ESP)on the structures considered(B3LYP/6-311++G(d,p)).
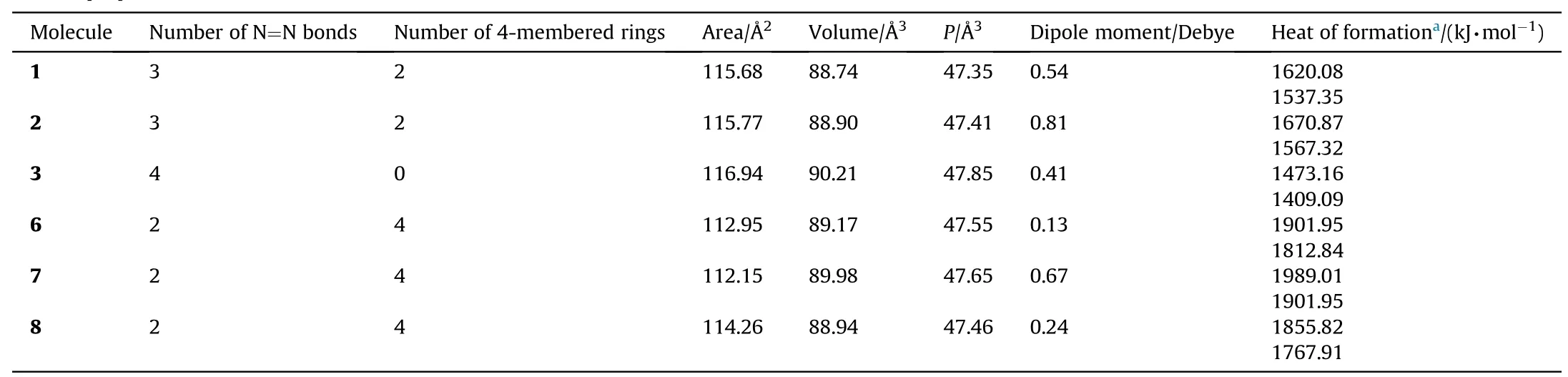
Table 1Various properties of the stable isomers considered.
Note that the heats of formation values in Table 1(f i rst entries)have been calculated by T1 recipe w hich closely reproduce heats of formation values calculated from G3(MP2).The later one has been developed for thermo chemical calculations.The T1 recipe operates by replacing the large basis set MP2 calculation by a dual basis set RI-MP2 calculation and replace the QCISD(T)calculation and vibrational frequency calculation by an empirical correction based on atom and bond counts[37].
All the structures in Table 1 are highly endothermic in nature.The T1 calculations yield the order of endothermicity as 7>6>8>2>1>3.The same order is produced by PM3 calculations based on B3LYP/6-311++G(d,p)geometry.Note that 6 has four 4-membered rings,w hereas 3 has none.Of the geometrical isomers 1 and 2,the later one(w hich has cis geometry)is more endothermic.As for 6,7,8 set of isomers(they all have the same number of 4-membered rings),repulsive interactions betw een the lonepairs are to be blamed for the endothermicity order among them(at least in part).Note that in structure-3(the least endothermic one)the lone pairs at the fusion points of the rings are in antiorientation.Obviously the ring strain contribution of 4-membered rings are also be blamed for highly endothermic nature of these structures.
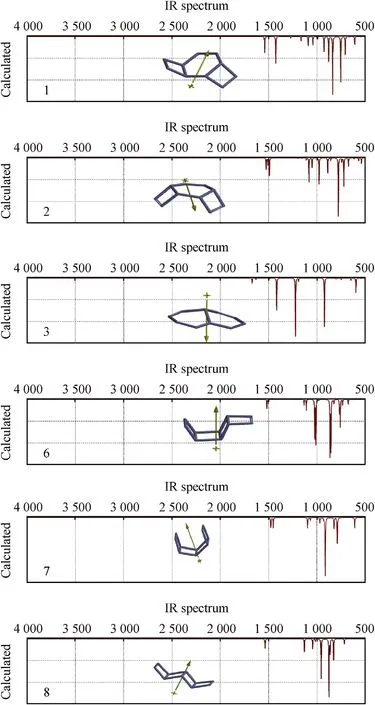
Fig.4.IRspectra of the stable structures considered(B3LYP/6-311++G(d,p)).
3.4.IR-spectra
Fig.4 show s the IR spectra of the stable isomers of present concern.Structure-3 w hich does not have any 4-membered rings has distinctly different IR spectrum than the others.The peaks at 1671 cm-1and 1631 cm-1stand for stretching of N=N bondsw hich are close to the site of positive end of the dipole moment vector.Whereas peaks at 1488 cm-1and 1415 cm-1are the stretching of N=N bonds w hich are close to the site of negative end of the dipole moment.The peak at the 1220 cm-1is stretching of N-N bonds linked to the fusion point.The peaks at 1189 cm-1and 1016 cm-1stand for stretching of N-N bond common to both of the rings.IR spectrum of structure-1 exhibits N=N bond stretchings in the 4-memebered rings at 1545 cm-1and 1539 cm-1.The double bond in the six-membered ring occurs at 1424 cm-1and coupled w ith NN stretching of the bond in betw een 4-membered rings.
Structures 2,6-8 have N=N stretching around 1500 cm-1.The peaksabout 1100-700 cm-1are N-Nstretchings coupled w ith some bendings.
3.5.Energies
Tables 2 and 3 show various energies of the systems considered based on tw o different basis sets.
B3LYP/6-311++G(d,p)level of calculations yield the stability order as 5>4>9>3>1>2>8>6>7.Whereas B3LYP/cc-PVTZ level of calculations give the order as 5>4>3>1>2>8>6>7.Note that structure-9 failed to optimize at B3LYP/cc-PVTZlevel.Also note that the decomposed structures constitute the most and the next stable systems in the series and the remnants after decomposing is N6structure w hich is the stable“open book”form[27].The rest of the structures(stable structures)have the same stability order by the tw o calculation methods employed.
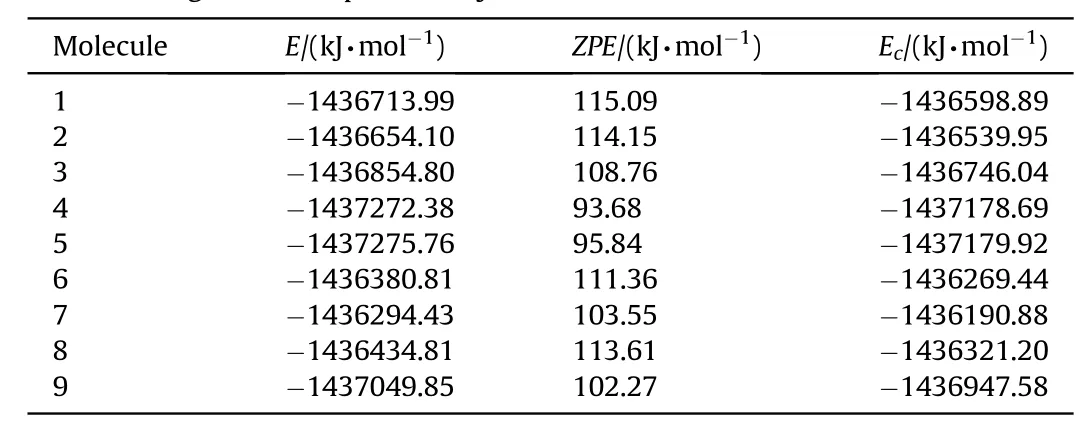
Table 2Various energies of the optimized systems considered.
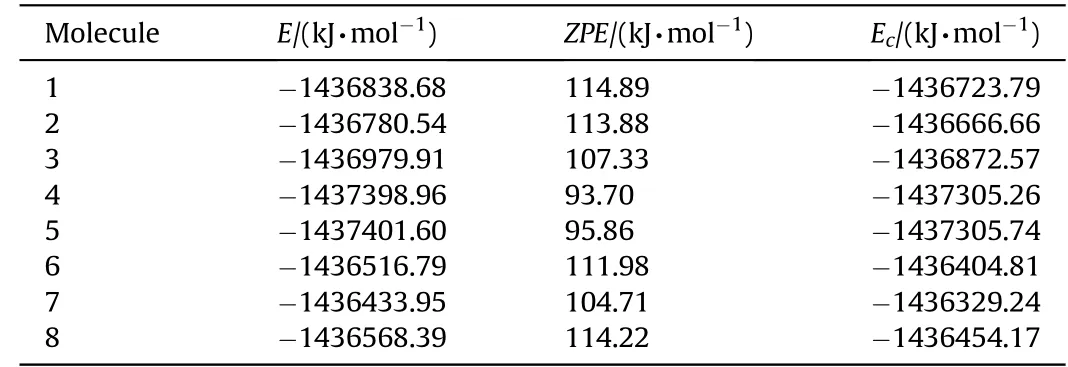
Table 3Various energies of the optimized systems considered.
Tables 4 and 5 displays the HOMO,LUMO energies and interfrontier molecular orbital energy gaps(Δε)of the structures considered.
B3LYP/6-311++G(d,p)level of calculations give the HOMO and LUMO energy orders as 4<5<8<7<1<6<9<2<3 and 3<7<9<6<8<4<1<5<2,respectively (see Table 4).Consequently,the HOMO-LUMOenergy gap,Δε,(interfrontier molecular orbital energy gap)has the order of 5>4>1>2>8>6>7>9>3.On the other hand,B3LYP/cc-PVTZ level of calculations yield HOMO and LUMO energy orders as 4<5<1<8<7<6<2<3 and 3<7<8<6<4<5<1<2,respectively(see Table 5).Whereas,the order ofΔεvalues is 5>4>1>2>8>6>7>3.Structure-3 has the highest HOMO energy w hereas 1 possesses the lowest among the stable isomers.As for the LUMO energies,structures-3 and 2 have the low est and highest energies,respectively.
The interfrontier molecular orbital energy(FMO gap)values for the stable isomers follow the same order by the tw o calculation methods employed.In each case,structure-3 has the smallestΔε value.The impact sensitivity is generally correlated w ith the HOMO-LUMO energy gap[40].Accordingly,structure-3 should be the most sensitive one to impact.
3.6.UV-VISspectra
Fig.5 show s the calculated UV-VIS spectra of the stable N10isomers.Structures 1,2(geometrical isomer of 1)and 8 all absorb in the UV-region.Structures-6 and 7 partly absorb also in the visible region.Spectrum of structure 3 spreads over UV region as w ell as visible region up to 550 nm Indeed the interfrontier molecular orbital gap(Δε)for 3 is the smallest of all(see Tables 4 and 5).
The possible conjugative effects betw een the nitrogen atoms in structure-3 narrow s the interfrontier energy gap(HOMO-LUMOenergy difference)thus causing a bathochromic effect as compared to spectra of the others.Structures 6 and 7 also have some absorption in the visible region.This might be due to through-space interaction betw een the N=Nπ-bond and the nitrogen lone-pair at the fusion points in 6 and through-space interaction betw een the N=Nπ-bonds w hich are absent in structure-8.
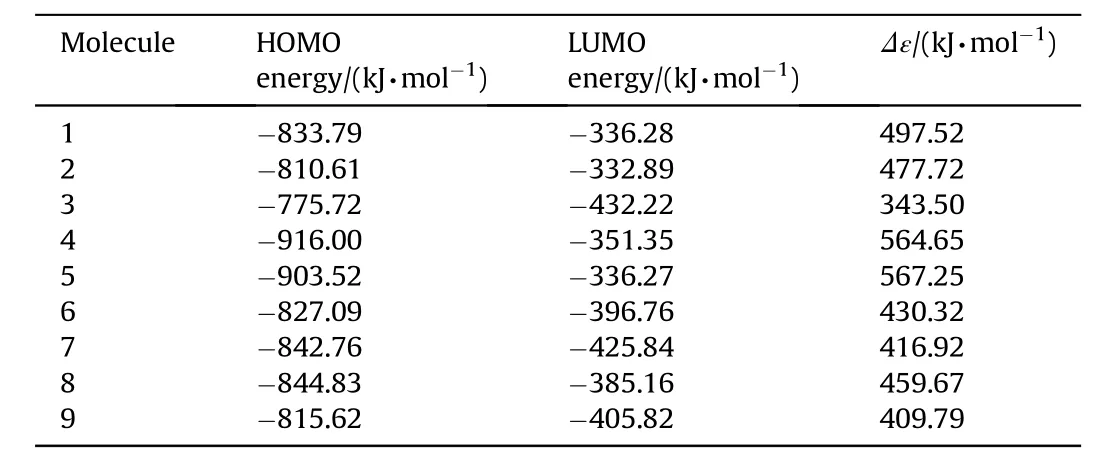
Table 4The HOMO,LUMO energies and interfrontier molecular orbital energy gaps(Δε)for the systems considered.
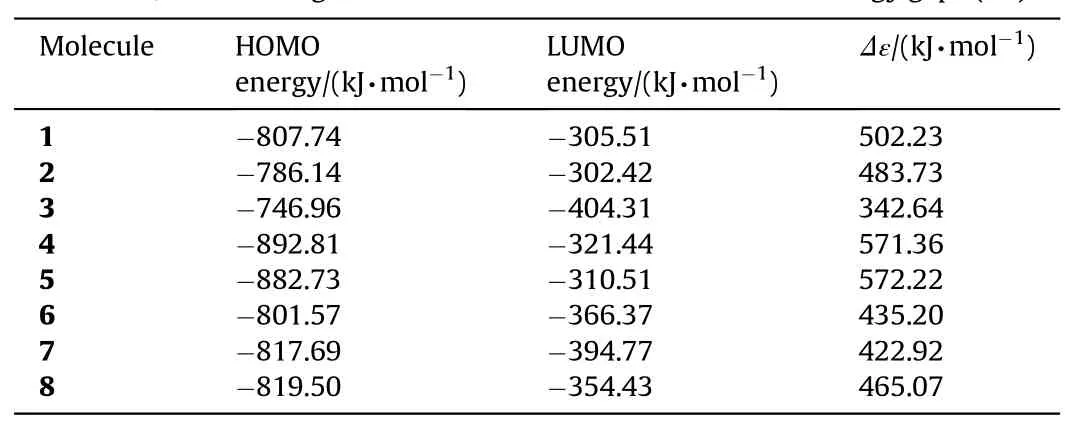
Table 5The HOMO,LUMO energies and interfrontier molecular orbital energy gaps(Δε).
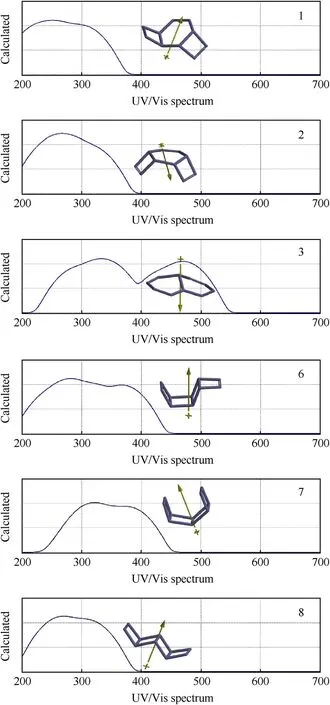
Fig.5.UV-VISspectra of the stable isomers considered(B3LYP/6-311++G(d,p)).
3.7.Impulse values of stable N10 structures considered
Green energetic materials,producing harmless waste products are favored due to obvious reasons.Polynitrogen compounds having high energy content are desirable materials.Polynitrogen compounds are believed that they w ill allow solid rocket propellants to compete in terms of energetic ef f i ciency w ith liquid propellants[2,18].For propellants,the material's potential is best measured by its speci f i c impulse,Isp.The speci f i c impulse in units of seconds can be approximated w ith the follow ing equation[5].
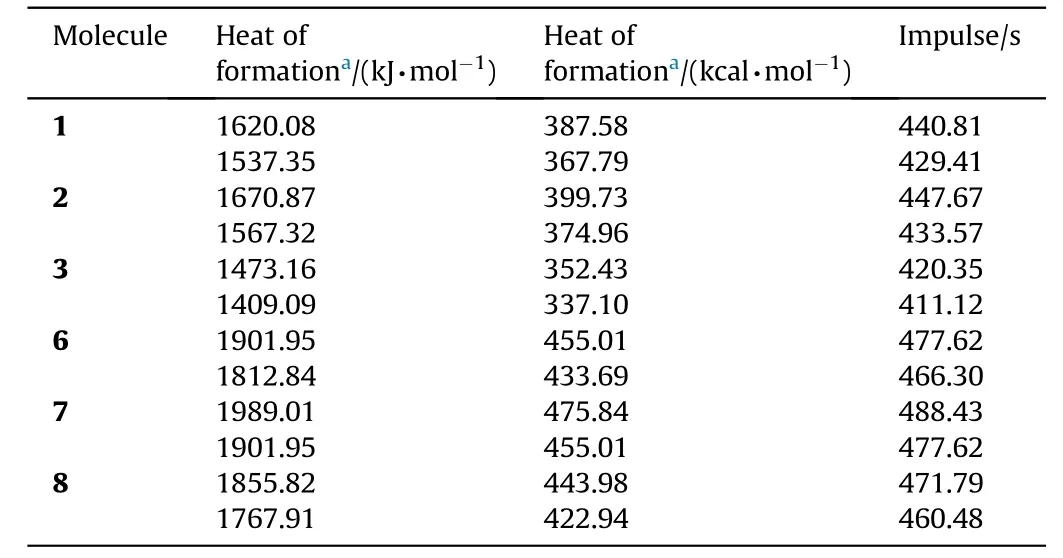
Table 6The heat of formation and speci f i c impulse values of the stable cyclic N10 structures.
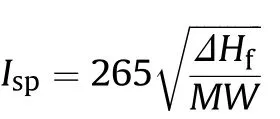
Where Ispin seconds,ΔHfin kcal/mol,MW in g/mol.
Presently that property has been estimated for the stable N10isomers(see Table 6).Both sets of heat of formation values yield the order of speci f i c impulse values as 7>6>8>2>1>3.Structure-7 having four 4-membered rings should have high strain energy,thus high heat of formation value and high speci f i c impulse.
4.Conclusion
Of the studied cyclic N10isomers,some of them w ere found to be stable(1-3,6-8)but highly endothermic(7>6>8>2>1>3)w ithin the restrictions of density functional theory and the basis sets employed(B3LYP/6-311++G(d,p)and B3LYP/cc-PVTZ,restricted closed-shell).Of the decomposed structures,4 and 5 eliminate tw o N2units to yield bicyclic N6structure(having tw o 4-membered fused rings)w hereas structure 9 liberates single N2unit to form bicyclic N8(having a 4-membered ring fused to a six-membered ring)as the remnant structure.Structure-3 has the smallest interfrontier molecular orbital energy gap.According to the general assumption that the impact sensitivity correlates w ith the HOMOLUMO energy gap,structure-3 should be the most sensitive one to impact.
The order of speci f i c impulse values has been found as 7>6>8>2>1>3.Structure-7 having four 4-membered rings should possess high strain energy,thus high heat of formation value and speci f i c impulse.
Polynitrogen compounds are recently on the focus of increased theoretical and synthetic attention.How ever,the synthesis of such compounds involve great practical dif f i culties.Nevertheless,progress in the synthesis of new polynitrogen compounds are highly aided by the theoretical methods in order to predict their properties etc.,prior to their synthesis.

- Defence Technology的其它文章
- Magnesium nanocomposites:An overview on time-dependent plastic(creep)deformation
- Ballistic performance of tungsten particle/metallic glass matrix composite long rod
- The detrimental effect of autofrettage on externally cracked modern tank gun barrels
- Information hiding w ith adaptive steganography based on novel fuzzy edge identi f i cation
- Optimization of gas tungsten arc w elding parameters for the dissimilar welding between AISI 304 and AISI 201 stainless steels
- Parametric study of single con f i ned fragment launch explosive device