
美国《Emerging Infectious Diseases》2019年第25卷第3期有关人兽共患病论文摘译
2019-05-23 03:41:08周银发
中国人兽共患病学报 2019年5期
P465 2015-2016年在印度尼西亚传播的禽流感A(H5N1)病毒重新分类//Desniwaty Karo-karo, Rogier Bodewes, Hendra Wibawa, 等
高致病性的禽流感(HPAI)A(H5N1)病毒从2003年开始在印度尼西亚(印尼)传播,主要危害家禽的健康,造成严重的经济损失以及导致168例实验室确诊的人类病例死亡。2015-2016年H5N1病毒在西爪哇省家禽间暴发,我们对在此期间收集到的39株H5N1病毒样本的全基因组进行系统进化分析,并与印尼近期公布的H5N1病毒序列进行比较。系统进化分析显示所有样本的血凝素基因都在2.3.2.1c支系的2个基因簇中。我们同时研究了这些基因簇中的神经氨酸酶、核蛋白、聚合酶以及碱性聚合酶1基因。一些高致病性禽流感病毒的基质、非结构蛋白和碱性聚合酶2基因,与2.1.3支系的亲缘关系更近而不是2.3.2.1c支系。并且有1例高致病性禽流感病毒的碱性聚合酶2基因与欧洲低致病性禽流感的亲缘关系非常近。我们在印度尼西亚流行的高致病性禽流感病毒中一共发现了13种重组类型,大部分发生在Indramayu市庭院饲养的鸡当中(图1)。
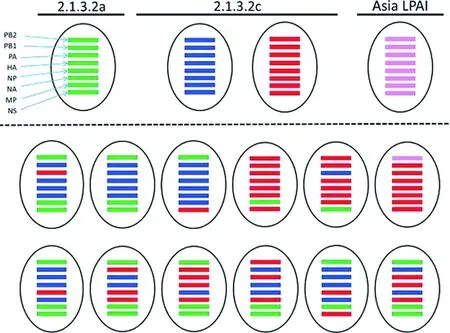
图1 2015-2016年印度尼西亚禽流感A(H5N1)病毒间重组情况,有些是通过最大似然比分析2.3.2.1c支系中的亲本毒株、2005-11(2.1.2.2a支系)和亚洲低致病性禽流感病毒进行确认。虚线以上的是亲本毒株,虚线以下的重组毒株。HA:血凝素;LPAI:低致病性禽流感;MP:基质蛋白;NA:神经氨酸苷酶;NP:核蛋白;NS:非结构蛋白;PA:聚合酶;PB1:碱性聚合酶1;PB2:碱性聚合酶2。绿色条杆代表2.1.3.1a支系;蓝色: 2.1.3.2c支系的A亚群;红色: 2.1.3.2c支系的B亚群;紫罗兰色:亚洲低致病性禽流感。
P482 基于单核苷酸多态性鉴定结核分枝杆菌复合群亚种及其相关谱系//Samuel Lipworth, Rana Jajou, Albert de Neeling, 等
结核病属于人兽共患病,我们对其临床表型及其造成的全球疾病负担认识不足并低估。这个问题在一定程度上是由于现有的实验室能力有限,无法利用计算机工具来准确鉴定结核分枝杆菌复合群(MTBC)的所有亚种。我们利用单核苷酸来识别结核病(SNP-IT),这是一种基于单核苷酸多态性来鉴定包括动物支系在内的所有结核分枝杆菌复合群成员。应用SNP-IT分析英国参比实验室收集的一系列临床基因组,我们检测到超出预期的大量的羚羊分枝杆菌分离株。羚羊分枝杆菌分离株的检出率与牛分枝杆菌分离株相似,然而在这之前羚羊分枝杆菌分离株在英国却从未被提及过。从国际视角来看,可能是因为羚羊分枝杆菌是种被低估的人兽共患病。准确的菌种鉴定能够使我们对结核分枝杆菌复合群所有亚种的临床表型、宿主范围以及传播机制进行更加细致的研究。
P489 应用基因组学研究新西兰历史输入性的产志贺毒素大肠埃希氏菌O26和非产毒变种//A.Springer Browne, Patrick J. Biggs, David A. Wilkinson, 等
产志贺毒素大肠埃希氏菌O26是一种重要的公共卫生病原体。一个国家的系统进化的细菌谱系与其进口活牛这个主要传染源的时间和数量有关。我们对新西兰的152株大肠埃希氏菌O26进行测序,并与来自其他14个国家的252株大肠埃希氏菌O26基因组进行比较。人类、动物源和食物源的菌株间基因变异与原产国及产志贺毒素密切相关,而与菌株分离源无关。对起源时间的估计表明从20世纪20年代到80年代,O26的21型菌株至少有3次输入到新西兰,而无毒力的O26的29型菌株在21世纪早期输入到新西兰。新西兰产志贺毒素大肠埃希氏菌O26的输入明显少于其他国家(如日本),这可能与活牛贸易模式有关。
P507 应用简化模型在无系统性分子流行病学项目的国家中开展结核病传播监测//Juan Domínguez,Fermín Acosta, Laura Pérez-Lago, 等
很多国家无法利用系统性分子或基因流行病学研究开展结核病监测。我们选择巴拿马作为简化模型替代策略的研究点。分枝杆菌分散重复单元-可变数目串联重复序列(MIRU-VNTR)分析显示,在巴拿马和科隆2个省有50%的结核分枝杆菌分离株包含在6个群簇(A-F)中。A群相当于北京基因亚型。全基因组测序(WGS)将由于近期活跃传播形成的低单核苷酸多态性的群簇(C群)与长期流行菌株形成的高多态性群簇(A、B群)区分开。在巴拿马有良好应用前景的3个特制的菌株特异性PCR,是以通过全基因组数据鉴定的单核苷酸多态性标记物为靶点,分析结果表明31.4%的病例与A-C群有关,北京基因型出现的概率较高且大部分集中在科隆省。合理的整合分散重复单元-可变数目串联重复序列分析、全基因组测序和特制的菌株特异性PCR可以成为无系统性分子流行病学项目的国家监测结核病的新模型。
P515 2017年欧洲人类阪崎肠杆菌感染情况的多中心研究//Sarah Lepuschitz, Werner Ruppitsch, Shiva Pekard-Amenitsch, 等
阪崎肠杆菌已经被记载为一种主要发生在新生儿当中且能够威胁生命的传染病病因。我们通过多中心研究(Multicenter Study)评估欧洲阪崎肠杆菌感染情况及暴发波及范围。要求欧洲24个国家代表的国家协调员将2017年收集的所有感染人类的阪崎肠杆菌菌株提交给澳大利亚的一所研究中心。在该中心开展的实验包括基质辅助激光解吸/电离飞行时间质谱法菌种鉴定、基于全基因组测序(WGS)分型,以及检测抗菌剂耐药性。11个国家共提交77株分离株,包括2017年收集的36株以及以往的41株。59株分离株通过WGS确认为阪崎肠杆菌,凸显出正确鉴别克罗诺杆菌属所面临的挑战。基于全基因组测序分型揭示了菌株高多样性,表明在2017年未发生跨国疫情暴发,但同时也确认了4起之前未公布过的历史性暴发。因此全基因组测序被推荐作为准确菌种鉴定、分型以及病原体检测的方法。
P523 2007-2017年美国德克萨斯州休斯顿市密切接触结核病的儿童窗口期预防//Andrea T. Cruz, Jeffrey R. Starke
在本次回顾性研究中,我们评估了2007-2017年密切接触成人结核病患者的5岁以下儿童窗口期预防的安全性和结核菌素试验(TST)阳转情况。本次研究纳入的儿童,其体格检查以及胸部X线检查均无明显异常,且结核感染检测结果均为阴性。共752位儿童(其中有41%儿童与传染源共同居住)在窗口期接受预防性治疗,通常采用直接面视下服用异烟肼预防治疗。所有儿童均未发生肝毒性不良反应以及结核病进展。37位儿童(4.9%)的TST转阳,这与传染源是否为儿童的父母有关(危险度OR值为 3.2, 95% 置信区间为1.2~8.2)。TST阳转情况与痰涂片结果、痰培养阳性以及是否与传染源同住无关。考虑到药物的安全性以及危险分层的困难,接受结核病窗口期预防的儿童密切接触者的纳入标准可以适当降低。
P547 2012-2016年突尼斯耐药结核分枝杆菌全基因组测序//Imen Bouzouita, Andrea Maurizio Cabibbe, Alberto Trovato, 等
为研究突尼斯耐药结核分枝杆菌传播情况,我们对2012-2016年期间收集的46株耐多药结核病分枝杆菌进行全基因组测序。核基因组多位点序列分型将30株(65.2%)菌株分成3个簇,表明存在广泛的近期传播且哈勒姆(Haarlem)基因型为优势基因型。全基因组测序有助于公共卫生署实施适当的控制措施提供依据。
P589 全基因组测序在2017-2018年法国结核病暴发调查中的应用//Charlotte Genestet, Caroline Tatai, Jean-Luc Berland,等
2017年6月-2018年4月期间,在法国 4个偏远城市的14位居民被确诊为感染北京基因型SIT1活动性结核病患者。全基因组测序显示这些患者属于同一传播链。基于全基因组测序的实验室调查能够即刻追踪相关病例来提高结核病控制水平。
猜你喜欢
天津农学院学报(2022年4期)2023-01-16 04:59:56
——以云南墨江自治县为例
贵州民族研究(2019年9期)2019-10-24 03:27:36
生物学杂志(2019年1期)2019-02-15 05:24:04
神州民俗(2018年9期)2018-11-21 11:10:02
流行色(2017年1期)2017-05-31 19:18:01
现代检验医学杂志(2016年4期)2016-11-15 02:00:58
新西部·中旬刊(2016年5期)2016-06-08 10:25:35
湖南畜牧兽医(2016年3期)2016-06-05 08:37:56
兽医导刊(2016年12期)2016-05-17 03:51:42
当代畜禽养殖业(2014年7期)2014-02-27 07:59:20
