
原发性免疫缺陷病患儿临床特点及预后分析
2019-05-22 02:10:08冯媛南楠李小青张翠
中国全科医学 2019年14期
冯媛,南楠,李小青,张翠
原发性免疫缺陷病(primary immunodeficiency disease,PID)是一组以单基因遗传变异为主的罕见疾病,因基因突变使得免疫细胞和免疫分子发生缺陷引起的免疫反应缺如或降低,导致机体抗感染免疫功能低下的一组临床综合征。PID属罕见病,迄今发现290多种,其中已经明确致病基因300余种[1]。PID主要临床特点是自幼反复感染、易患自身免疫性疾病和恶性肿瘤。由于PID的临床表现极为复杂,我国目前仅有极少部分PID患儿得到确诊,故早期诊断和合理治疗是控制患病率、降低病死率、提高生活质量的关键。本研究就近3年余来收治的37例PID患儿临床资料进行分析总结,以期为基层临床儿科医师提供参考。
1 资料与方法
1.1 临床资料 选取2013年6月—2017年2月西安交通大学医学院附属儿童医院因反复感染入院后检测血常规、体液免疫功能及T、B淋巴细胞、自然杀伤(NK)细胞免疫分型,从中初步筛选出37例PID患儿。诊断及分类按照2011年[2]及2015年国际免疫学会联合会(IUIS)PID专家委员会(PID EC)最新分类标准[1]。37例患儿中临床诊断X连锁无丙种球蛋白血症(XLA)17例、慢性肉芽肿病(CGD)4例、X连锁高免疫球蛋白M综合征(XHIM)4例、湿疹血小板减少伴免疫缺陷综合征(WAS)4例、选择性免疫球蛋白A缺乏症(sIgAD)3例、联合免疫缺陷病(SCID)2例、可疑中性粒细胞功能缺陷2例、先天性胸腺发育不全(DGS)1例。37例PID患儿除3例sIgAD、1例SCID及2例可疑中性粒细胞功能缺陷患儿外,其余均经基因检测确诊。患儿均除外近期使用过激素、免疫调节剂等。
1.2 方法
1.2.1 免疫球蛋白(Ig)含量测定 取静脉血2.5 ml于干燥试管中,待完全凝固后,以3 400 r/min离心5 min(离心半径18.8 cm),取1 ml血清置于温浴槽(温度37 ℃)中温浴60 min,抽取温浴后血清200 μl在贝克曼IMMAGE Immunochemistry System全自动生化分析仪(双光径免疫浊度分析仪)上(所用试剂由德赛集团有限公司提供),以速率散射比浊法测定血清IgG、IgA、IgM含量。
1.2.2 T、B淋巴细胞、NK细胞计数测定 取静脉血2.5 ml于乙二胺四乙酸(EDTA)抗凝试管中,取全血样本50 μl常温下避光孵育相关单克隆抗体15 min,加溶血素,洗涤后室温下避光孵育荧光染料结合的羊抗小鼠抗体15 min(所用试剂均购自美国BD公司),洗涤后上机采用FACSDiva软件,结果分析采用FCS Express。流式细胞仪的使用参照标准程序。
1.2.3 基因检测 2013—2015年因本院检验条件有限,临床怀疑PID患儿建议重庆儿童医院或上海儿童医学中心完善相关基因检测。2016年后本院与迈基诺公司联系,通过高通量二代测序行PID相关基因检测。
1.2.4 治疗 37例PID患儿入院时均有明显的感染,多为细菌感染,WAS及SCID患儿易有病毒感染,所有患儿使用抗生素治疗,对于存在EB病毒或巨细胞病毒感染的患儿加用更昔洛韦或阿昔洛韦抗病毒治疗,除sIgAD患儿,余抗体缺陷患儿给予静脉注射用丙种球蛋白替代治疗。
1.3 观察指标 记录37例PID患儿的基本资料、主要临床表现、实验室检查结果、基因检测结果及预后。
2 结果
2.1 患儿基本资料 37例PID患儿中2例sIgAD为女性,其余35例均为男性;年龄54 d~10岁9个月,发病至确诊时间6个月~8年。其中4例有可疑家族史(1例兄长病毒性脑炎后遗症卧床5年,1例兄长出生后18 d夭折,1例有兄妹3人夭折,1例兄长感染后夭折)。37例PID患儿中抗体缺陷24例(64.9%),吞噬细胞数目、功能缺陷6例(16.2%),有明确综合征5例(13.5%),联合免疫缺陷病2例(5.4%)。
2.2 主要临床表现 37例PID患儿均有反复感染病史,感染部位主要为呼吸道、消化道。17例XLA患儿多为细菌感染引起的呼吸道感染,1例曾患脓毒性休克,1例合并化脓性脑膜炎;4例CGD患儿起病年龄均为新生儿期,生后均接种卡介苗后出现卡介苗反应,均有出生后肛周脓肿表现,2例有肺结核;4例XHIM患儿中3例有反复口腔溃疡;4例WAS患儿中1例因巨细胞感染出现间质性肺炎(见表1)。

表1 PID患儿主要感染情况(例)Table 1 Infection status of children with PID
2.3 实验室检查结果 治疗前,17例XLA患儿体液免疫IgG、IgA、IgM含量降低,B淋巴细胞计数明显降低;4例XHIM患儿中,3例IgM含量明显升高,1例正常,4例IgG、IgA含量不同程度降低,外周血中性粒细胞计数均〈1.0×109/L;2例SCID患儿体液免疫IgG、IgA、IgM含量及T淋巴细胞计数均降低。1例DGS患儿行X线片检查未见胸腺影。PID患儿主要实验室指标检查结果见表2。
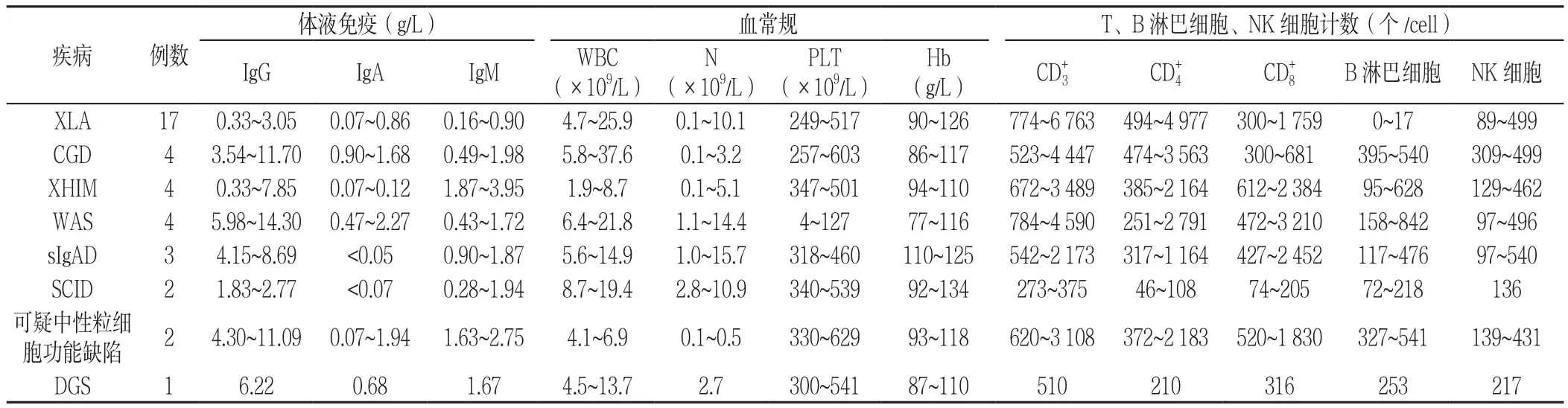
表2 PID患儿主要实验室指标(治疗前及治疗后多次检查结果)Table 2 Laboratory indexes of children with PID(results of multiple examinations before and after treatment)
2.4 基因检测结果 37例PID患儿除3例sIgAD、1例SCID及2例可疑中性粒细胞功能缺陷患儿外,其余均经基因检测确诊。17例XLA患儿基因检测结果均为BTK基因突变或变异;4例CGD患儿均为外院行呼吸爆发试验阳性;4例WAS患儿基因结果均为WASP基因突变;4例XHIM患儿基因突变均为CD40L突变,其中包括半合子突变3个,纯合突变1个;1例sIgAD及1例DGS患儿未收集到基因检测结果。
2.5 预后 随访1~5年,1例XLA患儿后期反复查血常规三系均降低,双下肢可见瘀斑及鼻衄表现,建议行骨髓穿刺,家属拒绝后失联;1例SCID患儿因重症感染死亡;1例XHIM患儿进行干细胞移植后失访。其余患儿均存活。
3 讨论
PID是指因遗传因素致免疫活性细胞和免疫活性分子发生缺陷引起的免疫反应缺如或降低,导致机体抗感染免疫功能低下的一组临床综合征,其共同特点为反复、严重、持续的感染,但病因不同,又有其各自的特点。每种PID的确切发病率不清楚。2015年IUIS PID EC最新分类标准将PID分为9大类,共涉及300多种基因突变导致的290余种PID:(1)T淋巴细胞、B淋巴细胞联合免疫缺陷;(2)其他已明确表型的免疫缺陷综合征;(3)抗体免疫缺陷病;(4)免疫失调性疾病;(5)先天性吞噬细胞数目、功能缺陷;(6)天然免疫缺陷;(7)自身炎症性疾病;(8)补体缺陷;(9)自身抗体相关的拟表型PID[1]。PID典型临床表现为反复感染,容易合并自身免疫性疾病及肿瘤性疾病[3]。本研究显示,PID自发病至确诊时间跨度短则月余,最长达8年。COOPER等[4]研究发现,PID患者通常从症状出现到被确诊之间的时间跨度平均长达9.2年,同时,COOPER等[4]转载了PID 10大临床预警症状:(1)1年内中耳感染次数〉4次;(2)1年内严重鼻窦感染次数〉2次;(3)抗生素治疗2个月疗效不佳;(4)1年内患肺炎次数〉2次;(5)婴幼儿体质量不增或生长发育极度迟缓;(6)反复深部皮肤或器官脓肿;(7)持续鹅口疮或皮肤真菌感染;(8)需要静脉应用抗生素以清除感染灶;(9)≥2处的顽固性感染(包括败血症);(10)有PID家族史。如果患儿临床具备≥2条提示临床医生应警惕PID的发生。中华医学会儿科学分会免疫学组提出规范PID临床疑似诊断路径、完善实验室建设,及时地发现PID患者,从而改善患者的远期疗效和生活质量问题[5]。
由于PID临床表型极为复杂,确诊有赖于免疫学筛查和基因分析,为了提高我国PID诊治水平,中华医学会儿科学分会免疫学组在全国范围内开展PID的全国协作研究,提出XLA、CGD、XHIM、WAS等较常见PID的初步诊断及筛查方法[5]。
XLA是由于人类BTK基因突变,B淋巴细胞发育障碍引起的PID。本组17例XLA患儿均为男性,均通过基因检测确诊。XLA在确诊前常见的临床表现为呼吸系统、皮肤软组织或深部位化脓菌感染,其中以肺炎多见,常见为细菌感染(肺炎链球菌、流感嗜血杆菌、肺炎支原体、脑膜炎球菌、革兰阴性菌、肠道病毒及蓝氏贾第鞭毛虫)[6]。预防接种中,IgG缺乏患儿不建议接种活疫苗,在接受免疫球蛋白替代治疗过程中,不建议接种减毒活疫苗(除流感疫苗外)[7]。本组患儿实验室检查结果可见,血清IgG、IgA、IgM含量降低,同时B淋巴细胞数量明显降低。因此,临床符合如下诊断要点,需警惕XLA:男性,反复感染,感染部位以呼吸道多见,常为化脓性细菌感染。突出临床体征是浅表淋巴结和扁桃体很小或难触及。血清IgG、IgA、IgM含量均降低,总Ig〈2.50 g/L,外周血B淋巴细胞明显减少〈2%。BTK基因分析可明确诊断,母系家族中男性可有类似病史。免疫球蛋白替代治疗可控制大多数XLA的感染症状及全身症状。
在PID中,以sIgAD最常见[8]652-653。本组资料中3例sIgAD均为临床诊断。发病年龄4个月~3岁,既往均有反复呼吸道感染病史,当地每次抗感染治疗有效,均因肺炎在本院后常规检查发现IgA含量极低,〈0.05 g/L。因此,sIgAD临床表现为反复呼吸道或胃肠道感染,常伴有其他系统疾病,如自身免疫系统疾病、过敏性疾病及恶性肿瘤等,实验室指标提示血清IgA〈0.05 g/L,而IgG和IgM含量正常或增高,细胞免疫功能正常,尚需除外其他因素如药物、脾切除术等致继发性IgA低下者。本病尽量避免使用血液制品,如静脉注射用丙种球蛋白,因其仅含微量IgA,不能选择性替代IgA从而达到预防作用,更重要的是重复应用可能导致过敏反应的严重并发症。对于sIgAD的治疗主要是治疗各种伴发疾病。
SCID为一组主要表现为T淋巴细胞缺陷,同时伴有其他细胞缺陷的异质性疾病,目前发现至少18种不同基因的突变所导致的疾病。包括XLA、Jak3缺陷、腺苷脱氨酶缺陷等病情严重。本组资料中2例患儿均为重症SCID,1例经基因检测确诊,最终因重症感染死亡,1例未行基因检测,后失联,实验室检查可发现绝大部分患儿外周血T淋巴细胞和NK细胞显著减少,B淋巴细胞显著增高,Ig含量多全面降低,重症SCID对所有病原体普遍易感,包括常见细菌、分枝杆菌、耶氏肺孢子菌、人巨细胞病毒、EB病毒、弓形虫及隐孢子虫等[9]。本病唯一的根治办法为造血干细胞移植。
伴有免疫缺陷的明确综合征是指具有特征性临床表现,同时伴有免疫缺陷的疾病。本组资料中包含4例WAS,1例DGS患儿。WAS是一种罕见的X-连锁隐性遗传性疾病,以血小板减少、血小板体积减小、湿疹、免疫缺陷、易患自身免疫性疾病和淋巴瘤为特征。如不经过造血干细胞移植,WAS蛋白表达阴性患儿生存期仅约为15岁[10]。WAS患儿易出现反复化脓性中耳炎、肺炎及皮肤感染,对卡氏肺孢子菌(PCP)、巨细胞病毒、单纯疱疹病毒(HSV)、EB病毒和腺病毒等易感。建议严格避免接种活病毒疫苗及接触水痘-带状疱疹病毒(VZV)、HSV等。所有WAS患儿推荐定期应用免疫球蛋白替代治疗(不考虑目前IgG水平),急性感染时可增加免疫球蛋白剂量。DGS又名先天性胸腺发育不全,患儿表现为特殊面孔,如眼距增宽、耳位低且有切迹、上唇正中纵沟短小等。常存在大血管异常,如法洛四联症和主动脉弓右位等先天性心脏病及顽固的低血钙搐搦症。生后出现反复病毒真菌或PCP感染,或慢性感染过程。患者X线片检查无胸腺影。组织病理检查:淋巴结深皮质胸腺依赖区的淋巴细胞减少。
CGD是少见的原发性吞噬细胞免疫缺陷病、发病率为1/(200 000~250 000)[11],由于还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶缺陷,患者于儿童早起发生反复致命的细菌和真菌感染,在慢性炎症部位形成肉芽肿。男性多见,男女比为6∶1[8]658-670。本组资料中收录4例CGD患儿,均经基因检测确诊,均为男孩,发病年龄均为新生儿发病,均有接种卡介苗后出现卡介苗反应,且均有婴儿期肛周脓肿的表现。此后反复出现多脏器感染,感染重,抗感染治疗时间长。CGD患儿常见感染的病原菌为:过氧化氢酶阳性细菌和真菌,肺部感染时常见病原为曲霉菌(41%),金黄色葡萄球菌(12%),其他还包括分支杆菌等[12]。因此类患儿均反复重症感染,治疗中需要兼顾多种病原菌感染,花费大,住院时间长。因此,对于反复严重感染,外周血免疫细胞分型T、B淋巴细胞计数均正常,血清Ig含量正常或升高,硝基四氮唑蓝(NBT)还原试验阴性或者NADPH氧化酶活性降低者均应高度怀疑CGD,基因测序可助确诊。
高免疫球蛋白M综合征(HIM)是一组少见的免疫缺陷病,原发性HIM因遗传方式不同,分为X连锁和非X连锁,XHIM占70%,其发病机制为:CD40L基因突变的结果改变了CD40L蛋白的晶体结构,使其不能与CD40分子结合,导致T淋巴细胞依赖抗原的再次免疫应答障碍。患儿易发生细菌、PCP感染。主要临床表现有反复呼吸道感染,且易有口腔溃疡,〉50%的XHIM患儿有间断或持续性中性粒细胞计数减少[13]。本组资料4例XHIM患儿均有中性粒细胞计数减少及反复口腔溃疡,其中2例IgM含量升高,4例IgG、IgA含量明显降低,细胞免疫均未见明显异常。对于高IgM的治疗,可给予静脉注射用丙种球蛋白400~500 mg·kg-1·月-1,对于持续中性粒细胞计数减少可用粒细胞集落刺激因子(G-CSF)治疗。XHIM的预后较XLA差,自身免疫性疾病、肠道感染、中性粒细胞计数减速及恶性肿瘤的发生率很高,应及早为该类患者提供骨髓移植或干细胞移植[14]。
研究发现,儿童反复呼吸道感染病例中,PID占10%[15]。反复感染病史超过5年的PID患者,其慢性腹泻和中耳炎的发生率明显高于非PID患者。本组37例PID患儿临床表现均有反复感染病史,以呼吸道感染多见,绝大部分静脉用抗生素治疗有效,故对存在反复感染的疑似PID患儿,或有明确家族史、幼年起病、其他高度提示PID者,首先进行初步实验室筛查,如全血细胞计数和分类,血清Ig测定,T淋巴细胞亚群、B淋巴细胞和NK细胞计数及所占比例,并分析其临床意义。
综上所述,本研究通过对PID的临床特点及诊断要点进行阐述,以期提高临床医生,尤其是基层医生对该病的警觉性及初步认识,争取对该病做到早期诊断、早期干预、改善预后、提高生活质量。本研究中缺乏对患儿后续随访的记录,且对家族史追溯不详细,在今后的研究中会对PID患儿的家族史及诊断后的随访情况进行详细追踪随访,同时进一步对PID相关基因进行研究。
猜你喜欢
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
数学小灵通(1-2年级)(2021年11期)2021-12-02 01:30:20
天津医科大学学报(2021年4期)2021-08-21 02:14:34
中等数学(2020年8期)2020-11-26 08:05:58
小学生学习指导(低年级)(2020年4期)2020-06-02 09:09:26
数学小灵通·3-4年级(2017年11期)2017-11-29 01:35:42
中国卫生(2015年12期)2015-11-10 05:13:24
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:43
现代检验医学杂志(2015年6期)2015-02-06 01:44:07
中国医学科学院学报(2013年2期)2013-03-11 20:25:46
