
龟鹿二仙口服液质量标准研究△
2019-05-13 08:28:26史兆松马栋栋张淹李士栋周祥山
中国现代中药 2019年4期
史兆松,马栋栋,张淹,李士栋,周祥山
1.东阿阿胶股份有限公司/国家胶类中药工程技术研究中心/山东省胶类中药研究与开发重点实验室,山东 聊城 252201; 2.华润昂德生物药业有限公司,山东 聊城 252201
龟鹿二仙口服液由龟甲、鹿角、党参、枸杞子4味中药材组成。具有温肾益精的功效,临床上主要用于久病肾虚,腰酸膝软和遗精阳痿等。该产品质量标准中缺少鉴别和含量测定方法,为更好地控制产品质量,保证临床用药安全有效,对质量标准开展再提升研究。分别对龟甲、鹿角、党参、枸杞子四味中药材进行薄层色谱鉴别,对甜菜碱进行含量测定,为产品的质量控制提供数据参考。
1 材料
1.1 仪器
Ultimate3000型高效液相色谱仪(Dionex),UltiMate3000系列光电二极管阵列检测器,色谱柱为丙基酰胺键合硅胶为填充剂(150 mm×4.6 mm,5 μm);BS224S型电子天平(赛多利斯);C9960A型超声清洗仪(CBL Photoelectron Technology);薄层摄像仪(瑞士CAMAG公司);Mili-Qpius型超纯水制备仪(密理博公司);硅胶G薄层板(青岛海洋化工厂)。
1.2 试药
甘氨酸、甜菜碱、党参对照药材、枸杞子对照药材(中国食品药品检定研究院,批号分别为:110735-200102、110894-200503、121057-201206、121072-201109);各阴性对照样品均为自制;乙腈、甲醇均为色谱纯;水为纯化水,其余试剂均为分析纯。
龟鹿二仙口服液产自东阿阿胶股份有限公司,批号分别为:20140601、20140602、20140603、20140801、20150101、20150102、20150103、20150401、20150402、20150403。
2 薄层色谱鉴别
2.1 甘氨酸鉴别
取本品1 mL,置具塞试管中,加水2 mL,盐酸2 mL,密塞,置105 ℃干燥箱中加热6 h,取出,加水6 mL摇匀,滤液蒸干,残渣加10%乙醇10 mL使溶解,作为供试品溶液;取缺龟甲、鹿角的阴性样品同法制成阴性样品溶液。另取甘氨酸对照品,加10%乙醇溶解,制成质量浓度为1 mg·mL-1的溶液,作为对照品溶液。吸取供试品溶液及阴性对照溶液各2 μL、对照品溶液1 μL,分别点于同一硅胶G薄层板上,展开剂:苯酚-0.5%硼砂水溶液(4∶1),展开,取出,晾干,置105 ℃干燥箱中加热10 min,取出,立即喷以2%茚三酮乙醇溶液,至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。阴性对照色谱中无相应斑点,见图1。

注:1.甘氨酸;2~4.样品;5.阴性样品。图1 甘氨酸薄层色谱图
2.2 党参鉴别
取本品60 mL,加水饱和的正丁醇振摇提取3次(40、40、30 mL),合并正丁醇提取液,用氨试液洗涤2次,每次20 mL,弃去氨液,正丁醇液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。取缺党参的阴性样品同法制成阴性样品溶液。另取党参对照药材4 g,分别加水30 mL煎煮两次,每次30 min,合并煎液,滤过,滤液同法制成党参对照药材溶液。吸取上述3种溶液各20 μL,分别点于同一硅胶G薄层板上,展开剂:甲苯-乙酸乙酯-甲酸(15∶2∶2),展开,取出,晾干,喷以硫酸-乙醇(1∶1),置105 ℃干燥箱中加热约7 min,置紫外光灯(365 nm)下检视,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。阴性对照色谱中无相应斑点,见图2。
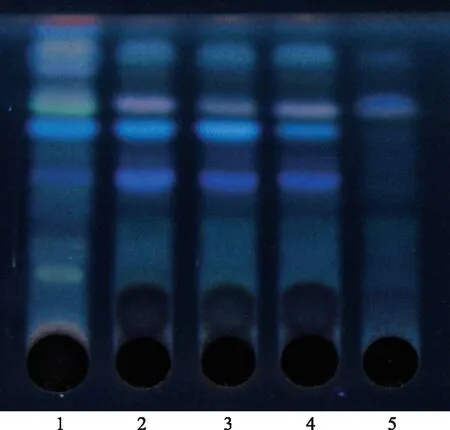
注:1.党参对照药材;2~4.样品;5.阴性样品。图2 党参薄层色谱图
2.3 枸杞子鉴别
取2.2项下制备的供试品溶液。取缺枸杞子的阴性样品和枸杞子对照药材2 g,同2.2项方法制成阴性样品和对照药材溶液。点样、展开、显色等操作同2.2项下方法。结果见图3。

注:1.枸杞子对照药材;2~4.样品;5.阴性样品。图3 枸杞子薄层色谱图
3 含量测定
3.1 色谱条件与系统适用性试验
采用HILIC(丙基酰胺键合硅胶)色谱柱(150 mm×4.6 mm,5 μm);以乙腈-水(80∶20)为流动相;柱温为30 ℃;蒸发光散射检测器检测漂移管温度为98 ℃;体积流速为0.8 mL·min-1。
3.2 甜菜碱对照品溶液
精密称取甜菜碱,加水溶解制成质量浓度为0.5 mg·mL-1的溶液,即得。
3.3 供试品溶液制备
精密吸取本品20 mL,置具塞锥形瓶中,精密加入80%甲醇40 mL,密塞,称定质量,超声处理(功率:800 W,频率:40 kHz)30 min,放冷,再称定质量,用80%甲醇补足减失的质量,摇匀,滤过,滤液蒸干,残渣加水10 mL使溶解,先上预处理的阴离子树脂柱(内径为1.5 cm,柱高为15 cm),用40 mL水洗脱,收集洗脱液,后上预处理的阳离子树脂柱(内径为1.5 cm,柱高为15 cm),用40 mL水洗脱至无色,弃去洗脱液,再用15%氨水溶液40 mL洗脱,收集洗脱液,蒸干,残渣加适量水溶解,转移至10 mL量瓶中,加水至刻度,摇匀,滤过,取续滤液,即得。
3.4 阴性样品溶液制备
精密吸取缺枸杞子的阴性样品20 mL,同3.3项方法制成。
3.5 专属性试验
分别精密吸取上述3种溶液各5 μL,按照3.1项下色谱条件进行测定。结果表明,阴性对照无干扰,见图4。理论板数按甜菜碱峰计算应不低于4000。
3.6 检测限及定量限
将甜菜碱对照品色谱峰测出的信号与空白样品测出的信号进行比较,分别以信噪比为3∶1及10∶1时注入液相色谱仪的量确定检测限与定量限,检测限为0.68 ng,定量限为2.40 ng。
3.7 线性关系考察
精密称取甜菜碱4.12 mg,置10 mL量瓶中,加水溶液至刻度,摇匀。分别吸取2.5、5、7.5、10、12.5、15 μL,按照3.1色谱条件依次进样,测定峰面积。以对照品的浓度为横坐标,峰面积积分值为纵坐标,得回归方程:Y=30.828X+23.654,r=0.999 9。甜菜碱在1.03~6.18 μg呈良好线性关系。
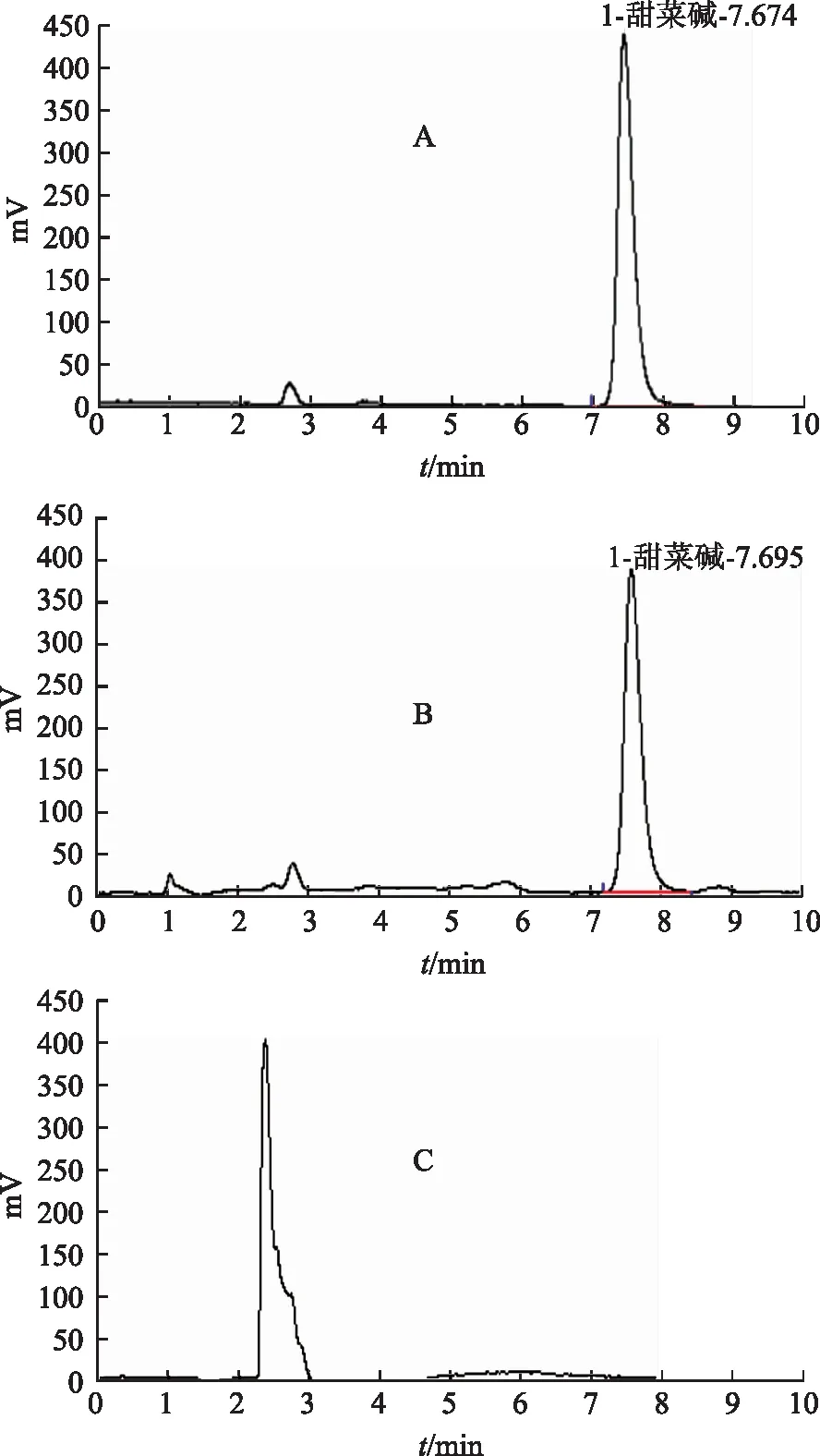
注:A.对照品;B.供试品;C.阴性对照样品。图4 甜菜碱HPLC图
3.8 精密度试验
取3.2项下对照品溶液,重复进样6次,每次10 μL,按照3.1色谱条件测定峰面积,计算RSD为1.47%(n=6),结果表明仪器精密度良好。
3.9 稳定性试验
精密量取本品(批号:20140801)20 mL,按照3.3项方法制备,分别于0、2、4、8、12、24 h进样6次,每次进样5 μL,测得峰面积,计算RSD为2.38%,结果表明,供试品溶液在24 h内稳定性良好。
3.10 重复性试验
精密量取本品(批号:20140801)6份,按照3.3项方法平行制备,按照3.1项色谱条件,分别进样5 μL测定,甜菜碱平均含量为0.226 mg·mL-1,RSD为1.07%(n=6),结果表明重复性良好,方法可行。
3.11 加样回收率试验
精密量取质量浓度为0.225 5 mg·mL-1的甜菜碱样品(批号:20140801)共6份,每份10 mL,分别精密加入甜菜碱对照品溶液(质量浓度为0.488 mg·mL-1)各6 mL,再加水至20 mL,制备6份供试品溶液,按照3.1项色谱条件,分别进样5 μL测定,计算回收率及RSD,结果见表1。
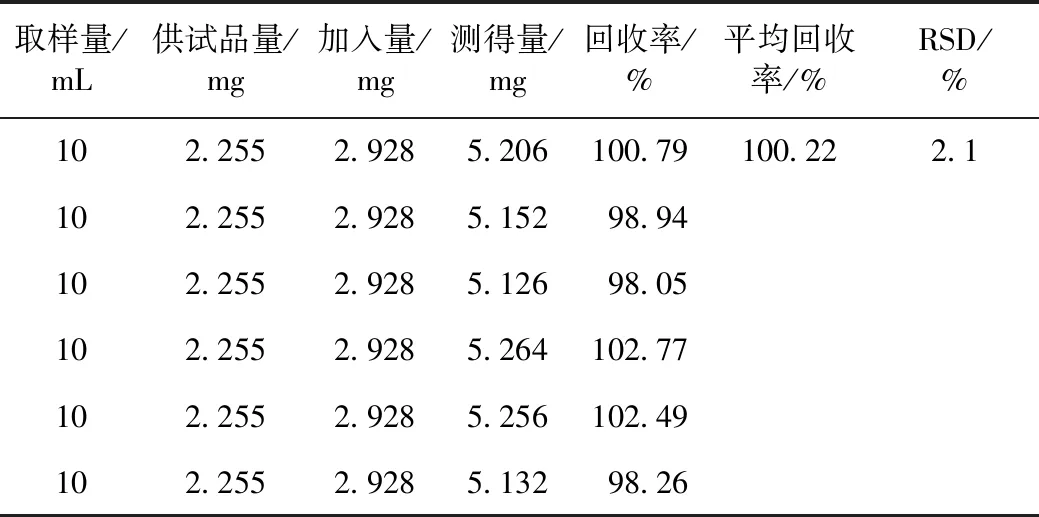
表1 加样回收率试验结果(n=6)
3.12 耐用性试验
取本品按照3.1项下方法试验,因甜菜碱为水溶性生物碱成分,很难在C18柱上有所保留,并且无特异的紫外吸收,故确定选用HILIC色谱柱按照3.1项下的色谱条件对其进行测定,含量测定结果无明显差别。
3.13 样品测定
精密量取10批龟鹿二仙口服液样品(批号分别为:20140601、20140602、20140603、20140801、20150101、20150102、20150103、20150401、20150402、20150403),按照3.3项下方法制备供试品溶液,按照3.1项色谱条件,分别进样5 μL测定,结果见表2。
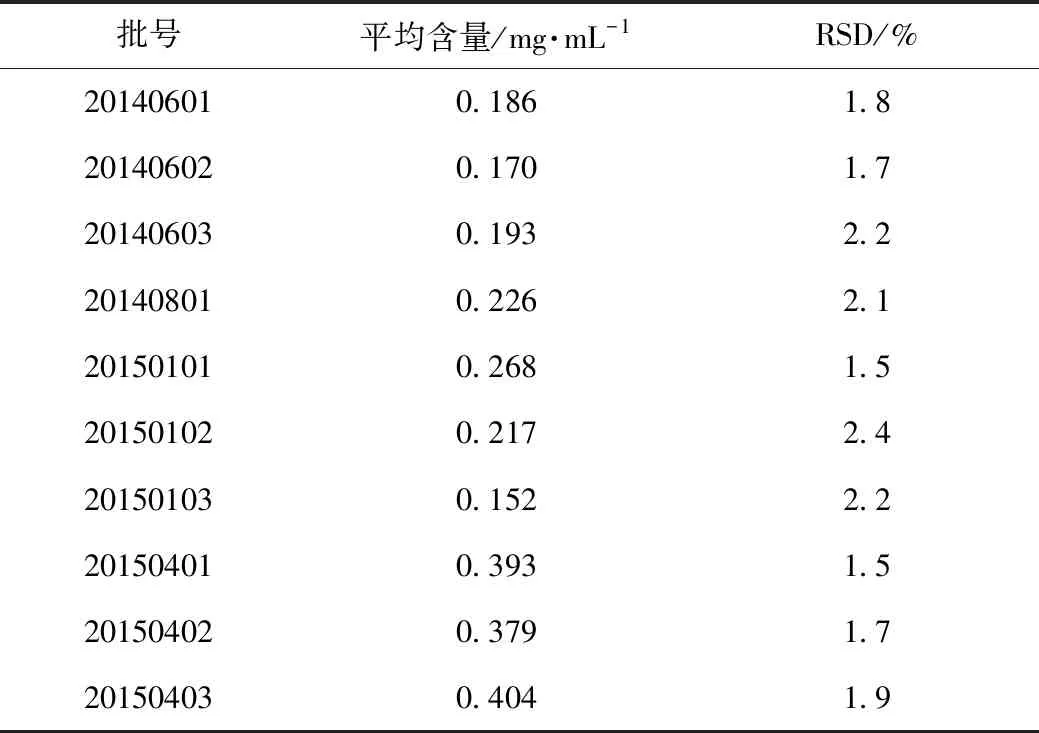
表2 样品测定结果(n=10)
4 讨论
龟鹿二仙口服液是由龟甲、鹿角、党参、枸杞子4味中药材组成的中药复方制剂,本研究通过色谱分离技术对处方中主要成分进行定性、定量分析。进一步提升了龟鹿二仙口服液质控水平,由于处方中成分复杂,分离困难、干扰多,通过上阴、阳离子树脂柱进行洗脱,除去氨基酸、蛋白质、多糖等成分,达到纯化供试品溶液的目的,使含量测定结果准确可靠。
原标准中仅采用定氮法测总氮量,专属性不强、产品质量标准较低。通过对处方成分分析,对产品中枸杞子有效成分之一甜菜碱进行研究,建立了HPLC含量测定方法。甜菜碱系甘氨酸的三甲基衍生物,季胺型水溶性生物碱,基本结构为(CH3)3-N+-CH2-COO-,分子式为C5Hl1N02,分子质量为117.11,无共轭结构。纯化方法曾采用0.1 mol·L-1异硫氰酸苯酯(PITC)乙腈溶液及1 mol·L-1三乙胺的乙腈溶液衍生后测定,发现衍生后未检测出理想甜菜碱成分峰;样品采用单阳离子柱洗脱后,测得成分峰很多且有重叠,甜菜碱成分分离度不理想。供试品选用上阴、阳离子柱洗脱处理后,杂质成分较少,没有出现干扰峰,且甜菜碱成分峰分离度较好。
本文对处方中全部药味均进行了薄层鉴别,在甘氨酸鉴别中,最初采用正丁醇-乙酸-水(3∶1∶1)为展开剂,但存在拖尾及背景干扰等缺点,经摸索,以苯酚-0.5%硼砂水溶液(4∶1)为展开剂时,分离度较好,展开效果理想,且在105 ℃加热10 min再喷显色剂比晾干后显色要好;在显色剂方面,2%茚三酮乙醇溶液比0.2%茚三酮乙醇溶液效果更明显。对党参、枸杞子鉴别中,分别选择各自对照药材作对照,在本实验选择确定的薄层色谱条件下,斑点清晰,分离度好,阴性对照无干扰。
龟鹿二仙口服液选取甜菜碱作为控制制剂质量的首选目标,根据《中华人民共和国药典》2015年版规定[1],枸杞子药材按干燥品计算,含甜菜碱不得少于0.30%,依据龟鹿二仙口服液处方,建议含量测定限度为不得少于0.15 mg·mL-1。
猜你喜欢
今日中国·西班牙文版(2023年2期)2023-02-14 03:05:44
文萃报·周二版(2020年22期)2020-06-08 10:52:31
宝藏(2019年5期)2019-06-21 01:23:28
云南中医学院学报(2015年2期)2015-07-31 18:11:59
中国洗涤用品工业(2015年8期)2015-02-28 19:02:48
保健与生活(2014年1期)2014-04-29 11:31:23
无机化学学报(2014年9期)2014-02-28 17:33:08
启迪与智慧·教育版(2013年6期)2013-04-29 00:43:54
化学分析计量(2013年5期)2013-03-11 16:37:48
中国洗涤用品工业(2012年4期)2012-03-20 15:39:33
