
立方纳米结构ZnGa2O4的制备及光催化性质
2019-05-10 06:54姜雁博矫淑杰高世勇王东博王金忠
发光学报 2019年5期
姜雁博,矫淑杰,高世勇,王东博, 王金忠
(哈尔滨工业大学 材料科学与工程学院,黑龙江 哈尔滨 150001)
1 引 言
环境污染问题已成为威胁人类生存发展的全球性问题,水污染是其中一个较为严峻的问题。目前,用于纺织、印刷、制革等工业的有机染料已成为水污染的一个重要来源。据统计,全球每年生产的有机染料,约有1%~20%在合成和染色工艺中成为了工业废水,而有机染料在自然界中很难被降解,并且大多数有机染料具有毒性,严重危害人类健康[1-3]。人们已采用多种方法、技术进行染料废水的处理,高级氧化法是目前降解有机染料的一个研究热点,特别是半导体纳米材料光催化氧化法[4-7]。光催化可将有机污染物最终氧化为二氧化碳和水,可以降解用传统方法难以降解的结构复杂的污染物。其中最具代表性的是TiO2纳米粉体多相光催化,但TiO2纳米粉体容易团聚、难以回收再利用、光响应波段较窄、光生电子/空穴复合率也比较高,因此仍需不断开发新的半导体光催化材料[8-12]。
大多数光催化剂主要集中于具有d0电子构型的Ti4+、Zr5+、Nb5+、Ta5+基化合物,以及具有d10电子构型的In3+、Ga3+、Ge4+、Sn4+基P区金属化合物等,一般具有d10电子构型的化合物都会展现出优异的光催化性质[13],ZnGa2O4正是具有d10电子结构的P区三元金属化合物[14]。ZnGa2O4是一种宽带隙(4.4~5.2 eV)p型半导体[15],其价带和导带位置均符合光催化制太阳能燃料的条件,即可以利用太阳能分解水制氢[14]或还原二氧化碳[16],将太阳能以化学能的形式存储起来。
作为典型的尖晶石氧化物,ZnGa2O4属于立方晶系,面心立方点阵。ZnGa2O4纳米材料的化学稳定性和热稳定性都很高,在光催化[17-21]、发光器件[22-26]、锂电存储[27]、生物成像[28]等领域具有很大的应用潜能。ZnGa2O4粉末或薄膜材料的制备方法主要包括高温固相反应法、溶胶-凝胶法、脉冲激光沉积法、水热法、射频磁控溅射等。ZnGa2O4性能方面的研究内容主要包括发光特性、余辉特性、光催化性质、储锂性能等,其中光催化主要是光催化制太阳能燃料,有关ZnGa2O4光催化降解有机污染物的报道并不多。半导体纳米材料光催化降解有机污染物在环境问题的治理中,尤其是染料废水的处理中具有非常大的应用价值,各种形态的ZnGa2O4纳米材料光催化降解有机污染物的活性有待我们去研究。
Liang等[29]采用两步水热法在FTO衬底上生长出多孔的ZnGa2O4微方块薄膜,ZnGa2O4微方块是由α-GaOOH纳米柱经溶解再结晶转变而来的,但立方块表面较粗糙有孔洞,尺寸较大;Han等[27]经一步水热法成功制备出ZnGa2O4纳米立方块粉末,但不同反应时间下获得的样品的TEM表征结果显示ZnGa2O4纳米立方块也是由α-GaOOH纳米柱经溶解再结晶转变而来的。粉体催化剂在光催化实验中需要多次离心操作,难以回收再利用,并且在降解有机染料时如果用量较大会影响溶液的透光度,削弱光催化作用,因此ZnGa2O4纳米立方块/FTO结构更适合用作光催化剂。本文基于两步水热法于FTO衬底上制备出结晶性良好、尺寸较小、分布密集的ZnGa2O4立方纳米结构,再以所制备的样品作为光催化剂,用有机染料作为模拟污染物,在模拟太阳光的辐照下测试样品的光催化性能。
2 实 验
2.1 ZnGa2O4立方纳米结构的制备
2.1.1 水热法制备前驱体α-GaOOH纳米柱阵列
硝酸镓(Ga(NO3)3·XH2O)作为镓源,六次甲基四胺(HMT)作为碱源,衬底选用FTO透明导电玻璃。将已清洗的FTO衬底(1.1 cm × 2.8 cm)固定于反应釜的聚四氟乙烯内衬中,并保证导电面始终朝下。分别配置30 mL 0.06 mol/L的硝酸镓水溶液和15 mL 0.1 mol/L的HMT水溶液,吸取3 mL HMT溶液加入到所配置的硝酸镓溶液中,经磁力搅拌均匀后即得到反应液,再将反应液转移至聚四氟乙烯内衬中。密封后将水热反应釜放入恒温干燥箱内,水热反应温度为150 ℃,反应时间为12 h。待温度自然降至室温时取出样品,用无水乙醇、去离子水清洗后,用氮气吹干待用。
2.1.2 水热法制备ZnGa2O4立方纳米结构
所制得的α-GaOOH纳米柱/FTO结构作为衬底和前驱体,二水乙酸锌(Zn(CH3COO)2·2H2O)作为锌源,反应液即为锌源的水溶液。保持水热反应温度为185 ℃,反应时间为24 h,改变锌源的量和反应液体积可获得不同形貌的ZnGa2O4纳米结构。首先固定反应液的体积为25 mL,改变锌源的质量配置溶液如表1所示,所获得的样品标记为第一组样品;再固定锌源的质量为0.220 g,改变反应液体积如表2所示,所获得的样品标记为第二组样品。
表1 第一组样品(V反应液=25 mL)的水热生长条件
Tab.1 Hydrothermal growth conditions of the first group samples

样品编号m锌源/gC反应液/(mol·L-1)10.1100.0220.1650.0330.2200.0440.2740.0550.3290.06
表2 第二组样品(m锌源=0.220 g)的水热生长条件
Tab.2 Hydrothermal growth conditions of the second group samples
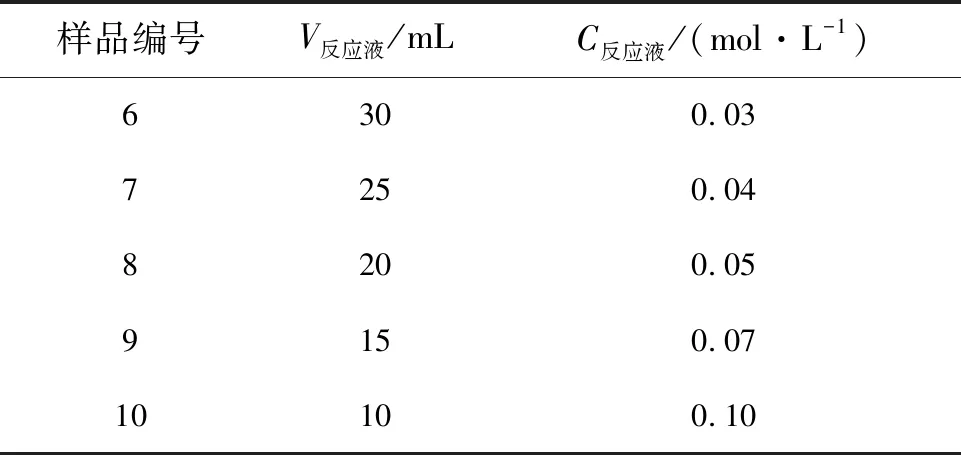
样品编号V反应液/mLC反应液/(mol·L-1)6300.037250.048200.059150.0710100.10
2.2 样品表征
所制备样品的晶相和晶体结构由X射线衍射仪表征(XRD,Panalytical/Empyrean),采用Cu Kα射线。形貌和尺寸由扫描电子显微镜(SEM, HRBNU/SU70)表征,加速电压为15 kV。紫外-可见分光光度计(*Phenix/UV1700PC)用来表征样品的吸收特性。
2.3 光催化性能研究
染料在光催化反应过程中的浓度变化可由λmax下的吸光度的变化来确定,其降解效率定义为:
(1)
C0是染料的初始浓度(5 mg/L),Ct是t时刻的染料的浓度;A0是染料在λmax下的初始吸光度,At是t时刻染料在λmax下的吸光度。
用于光催化实验的染料为罗丹明B(RhB)、亚甲基蓝(MB)、刚果红(CR)和甲基橙(MO),4种染料的水溶液的初始浓度均为5 mg/L。ZnGa2O4立方纳米结构(1.1 cm × 1.4 cm)作为光催化剂,采用氙灯(CEL-S500)作为模拟太阳光源。利用紫外分光光度计分别测试4种染料的紫外-可见吸收光谱,确定每种染料的最大吸收波长λmax及其在各自的λmax下的初始吸光度。在进行光催化实验时,先从4种染料中各吸取4 mL转移到4支石英试管中,再分别加入光催化剂,然后将石英试管置于磁力搅拌器上。在氙灯辐照下,每隔10 min对染料进行一次采样,测定每种染料在各自的λmax下的吸光度。在实验进行1 h后再一次测定各种染料的紫外-可见吸收光谱与初始吸光度进行比较。
3 结果与讨论
前驱体α-GaOOH纳米柱的结构与形貌可由XRD谱和SEM图进行分析,如图1(a) 所示,除去来自衬底FTO的衍射峰,所制前驱体的XRD衍射峰峰位和JCPDS: 06-0180相一致,表明所制前驱体是α-GaOOH,属于斜方晶系,晶胞参数为a=0.458 0 nm,b=0.980 0 nm,c=0.297 0 nm[20,27]。图1(b) 是α-GaOOH的SEM俯视图,低倍率SEM图显示获得了致密的α-GaOOH纳米柱阵列,由高倍率SEM图可知,纳米柱底面呈平行四边形,长边约250 nm,短边约200 nm。

图1 α-GaOOH纳米柱结构与形貌表征。(a)XRD谱(*用来标记来自衬底FTO的衍射峰;(b)SEM图。
Fig.1 Structure and morphology characterization of α-GaOOH nanorods. (a) XRD pattern(*mark the diffraction peaks from the FTO substrate). (b) SEM image.
图2是由第一组样品和第二组样品获得的具有代表性的SEM俯视图。图2(a)~(b)、图2(c)~(d) 和图2(e)~(f)分别为样品1、样品6和样品4的SEM照片。样品1中大部分立方块的边长接近1 μm,少数立方块的边长接近500 nm,还可观察到很多尺寸较小的纳米柱,这些纳米柱的形状、尺寸与图1(a)显示的α-GaOOH纳米柱相当。与实验条件对照表明用于制备样品1的锌源的质量偏少,还有很多α-GaOOH纳米柱未参与反应而暴露在衬底表面。反应过程中由于成核位点较少,立方块有足够的生长空间而具有较大的尺寸。在样品6的SEM照片中可观察到形状规则、尺寸均匀(约500 nm)、分布致密的立方块结构。与样品1对比,样品6中的立方块之间彼此交叠,还出现了六角纳米柱结构,由于用于制备样品6的锌源较多,表明样品中存在ZnO相(如红框标注部分);图2(e)、(f)给出了样品4的表面形貌,其中立方块的边长在500 nm左右,绝大多数立方块都沿着不同的方向交叠嵌套在一起。前驱体α-GaOOH纳米柱阵列的密度较大,在后期的反应中锌源充足,成核位点较多,立方块的生长空间受限而彼此嵌套。对比两组样品的形貌可以看出,随着锌源用量的增加或者反应液体积的减小,成核越来越容易进行,六角纳米柱结构逐渐消失,立方块密度和交叠程度都在随之增加。通过以上表征结果可知改变锌源用量可调制立方块纳米结构的尺寸,改变反应液体积可以控制立方块纳米结构的密度。

图2 ZnGa2O4立方结构的形貌表征。(a)、(b)分别为样品1的低倍和高倍SEM图;(c)、(d)分别为样品6的低倍和高倍SEM图;(e)、(f)分别为样品4的低倍和高倍SEM图。
Fig.2 Morphology of ZnGa2O4cubes. Sample 1 SEM images low(a) and high(b) magnification. Sample 6 SEM images low(c) and high(d) magnification. Sample 4 SEM images low(e) and high(f) magnification.
由图2中的SEM图可知样品4的立方结构形貌较为良好,尺寸比较均一,与之相对应的XRD图谱如图3(a) 所示。除去来自衬底FTO的衍射峰后,样品4的XRD衍射峰峰位和JCPDS: 38-1240相一致,表明所得立方结构为ZnGa2O4,属于立方晶系,晶胞参数为a=b=c=0.833 5 nm[20,27,29];由图3(b)所示的紫外-可见吸收光谱可知,样品4的吸收带落在紫外区,对于可见光的吸收很微弱。

图3 (a)基于XRD的ZnGa2O4立方纳米结构表征(*用来标记来自衬底FTO的衍射峰);(b)ZnGa2O4立方纳米结构的紫外-可见吸收光谱。
Fig.3 (a) Structure characterization of ZnGa2O4nanocubes based on XRD pattern(*mark the diffraction peaks from the FTO substrate). (b) UV-vis spectrum of ZnGa2O4nanocubes.
从图2可观察到,ZnGa2O4立方结构表面光滑、无孔洞,表明水热反应条件较为适宜,在晶相转变过程中纳米立方块无需通过产生孔洞来弥补体积变化,可以制备出结晶性较好的ZnGa2O4立方块结构。如图2(a)蓝框标注部分,由α-GaOOH纳米柱和ZnGa2O4纳米立方块共存可知,α-GaOOH纳米柱向ZnGa2O4纳米立方块的晶相转变是通过溶解再结晶实现的[27,29],反应方程如下:
α-GaOOH+H2O+OH-→[Ga(OH)4]-,
(2)
[Ga(OH)4]-+Zn2+→ZnGa2O4+H2O.
(3)
图4给出了ZnGa2O4纳米立方块的生长示意图,当锌源不足时,成核位点较少,得到体积较大的ZnGa2O4微方块;当锌源充足时,成核位点很多,生长空间受限,ZnGa2O4纳米立方块相互交叠,紧密分布于衬底表面。
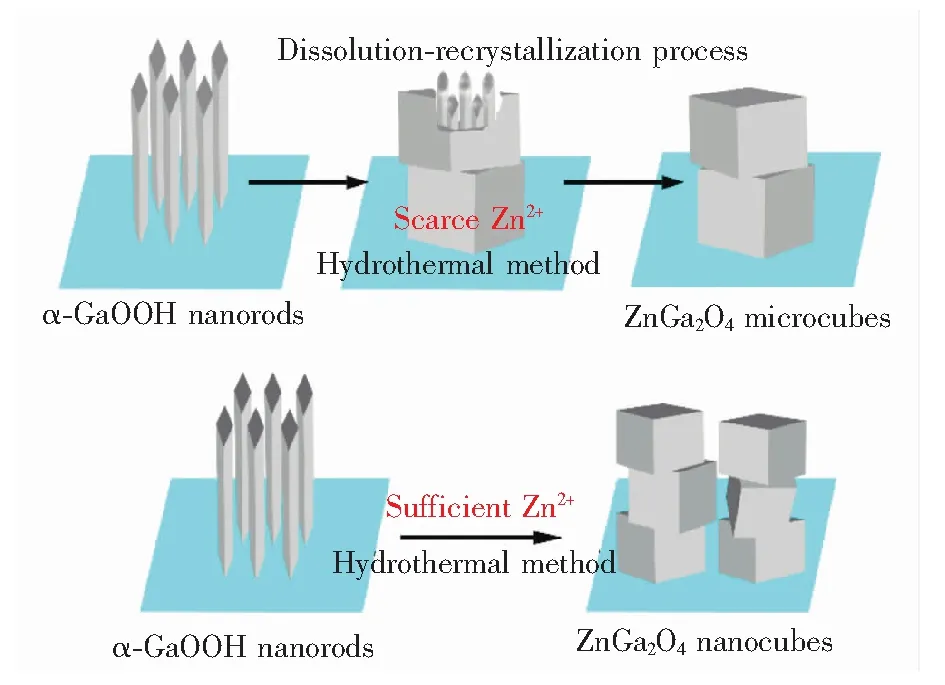
图4 从α-GaOOH纳米柱到ZnGa2O4立方块的晶相转变示意图
Fig.4 Schematic illustration of crystal phrase transition from α-GaOOH nanorods to ZnGa2O4cubes
利用样品4(1.1 cm × 1.4 cm)作为光催化剂进行光催化降解有机染料实验。降解前,所选染料的初始浓度均为5 mg/L,相应的紫外-可见吸收光谱如图5(a) 所示,MO的λmax=465 nm,相应的初始吸光度为0.322;MB的λmax=664 nm,相应的初始吸光度为0.427;CR的λmax=500 nm,相应的初始吸光度为0.25;RhB的λmax=554 nm,相应的初始吸光度为0.43。
图5(b) 给出了样品4对每种染料的降解效率随时间的变化规律。在前10 min内,MB降解效率可达22.8%,RhB、CR的降解效率分别达到4%、5.4%,而MO的降解效率仅为1.2%。10 min后RhB和CR的降解速率明显减慢并趋于稳定,MB染料的降解速率虽也有下降,但较之其他两种染料仍保持较高的降解效率。实验中还观察到光催化剂在降解MB、RhB和CR染料后均有轻微的颜色变化,表明光催化剂对这3种染料都有一定的吸附作用,前10 min内主要依靠吸附作用降低染料的浓度。10 min后催化剂对RhB、CR染料的吸附达到饱和,从而导致降解效率保持稳定,而随着时间的延长,光催化剂对MB染料仍具有持续的较高的降解能力。1 h后,MB的降解效率可达56%,RhB的降解效率为17.9%,CR的降解效率仅为9.5%。光催化剂对MO的降解行为显示了不同的特性,在实验时间内,虽然降解效率很低,但随着时间的延长,降解效率持续增加,最终效率达到15.6%,且光催化剂在降解MO后无颜色变化,表明光催化剂对MO的吸附作用很弱,实验中以光催化降解效应为主。MB、RhB、CR、MO在降解前、后的紫外-可见吸收光谱如图6所示,染料各自的λmax处的降解前后的吸光度的变化,可以直观地显示出光催化剂对每种染料的降解能力,这与图5(b)显示的结果一致。
由于实验中选用模拟太阳光作为光源,所以有机染料的降解由紫外光激发的光催化氧化过程和可见光激发的光敏化氧化过程共同完成[3,12],如图7所示。在紫外光辐照下,ZnGa2O4价带中的电子被激发到导带成为可自由移动的导带电子e-,而在价带中留下了可自由移动的空穴h+。迁移到催化剂表面的电子和表面吸附或溶解在染料中的氧分子反应,最终生成反应活性很高的·OH。表面的空穴和OH-或者水分子反应直接生成·OH;在可见光辐照下,催化剂表面附近的染料分子被激发到单态或者三重态,其电子注入到ZnGa2O4导带中,而自身成为阳离子自由基。迁移到催化剂表面的电子与氧分子结合,最后转变成·OH。·OH作为一种强氧化剂,可将绝大多数的染料分子最终氧化为二氧化碳和水。
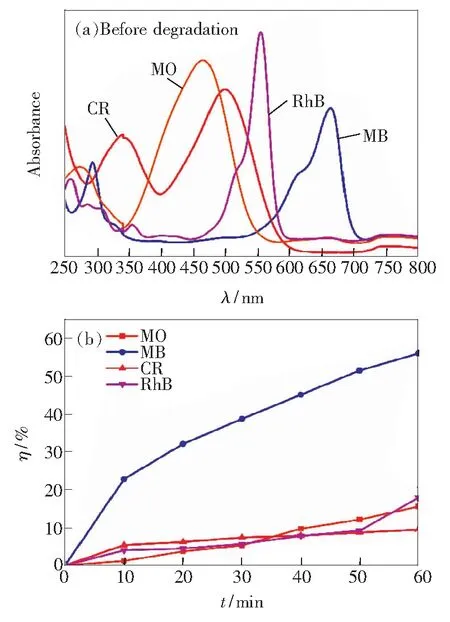
图5 (a)降解前4种染料(C0=5 mg/L)的紫外-可见吸收光谱;(b)氙灯辐照下ZnGa2O4立方纳米结构对每种染料的降解效率。
Fig.5 (a) UV-vis spectra of selected dyes(C0=5 mg/L)before degradation.(b)Degradation efficiency of dyes in the presence of catalyst under xenon lamp irradiation.

图6 4种染料降解前、后的紫外-可见吸收光谱。 (a)MB;(b)RhB;(c)CR;(d)MO。
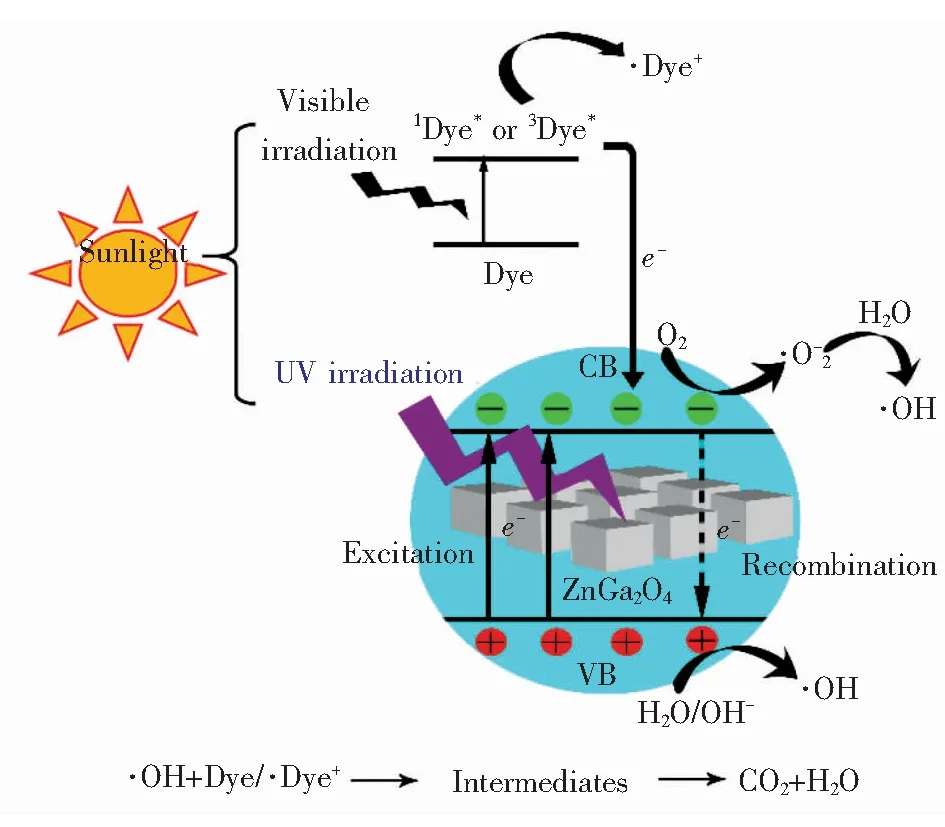
图7 ZnGa2O4立方纳米结构光催化降解有机染料机理图
Fig.7 Main mechanism for the photocatalytic degradation of organic dyes during ZnGa2O4nanocubes as the photocatalyst
由图3(b)的紫外-可见吸收光谱可知,ZnGa2O4的吸收带位于紫外区,对可见光的利用率极低,在太阳光照射下很难被激发产生光生电子-空穴对,所以实验中ZnGa2O4光催化降解有机染料主要由光敏化氧化过程实现,活性自由基主要由电子参与反应形成,而ZnGa2O4导带中的电子主要来自表面附近的激发态染料分子,因此染料分子自身的结构和性质是影响光催化活性的关键因素。由实验结果可知,ZnGa2O4立方纳米结构对4种染料各表现出不同的吸附能力和光催化活性,而吸附和光催化作用总是相辅相成的。催化剂对MB染料显示出较强的吸附能力和较高的光催化活性,对RhB、CR染料的吸附作用和光催化活性都很弱,对MO几乎不产生吸附,却显示出较弱的光催化作用。由于实验中反应体系处于缺氧环境,电子不能及时被氧分子捕捉,活性自由基很难生成,从而制约了样品的光催化活性。H2O2自身就是一种氧化剂,还可作为电子受体,为反应体系供氧。为了体现电子在染料降解过程中的作用,向两份4 mL、5 mg/L的MO染料溶液中各加入150 μL H2O2,磁力搅拌15 min后测量各溶液的吸光度,并以该吸光度值作为各自的初始吸光度。向其中一份MO溶液中加入样品进行光催化实验,同样,在氙灯辐照下,每隔10 min对没有催化剂的MO溶液也进行一次采样,得到两种实验条件下的MO染料的降解曲线如图8所示。在相同时间内,同时含有催化剂和双氧水的MO染料(标记为MOboth)的降解效率明显高于只加入双氧水的MO染料(标记为MOH2O2),并且随着时间的增加二者的差值越来越大。依据以下反应方程式:

(4)
(5)
(6)
(7)
(8)
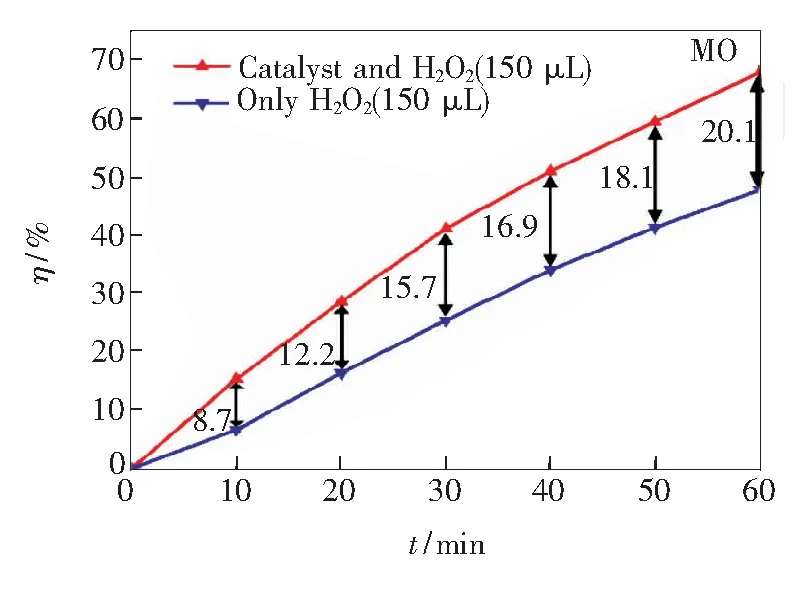
图8 MOboth和MOH2O2的降解曲线
(9)
相比于MOH2O2,MOboth中催化剂表面的电子可以和H2O2分解得到的氧分子结合最终生成·OH,也可以被H2O2捕获生成·OH,从而具有持续较高的降解效率。
4 结 论
本文首先采用水热法在FTO衬底上制备出底边长约250 nm的α-GaOOH纳米柱阵列,再经水热反应其将转变为ZnGa2O4立方纳米结构。当锌源用量偏少、浓度偏低时,边长约为1 μm的ZnGa2O4微方块松散地分布于衬底表面;当锌源充足、浓度较高时,边长约为500 nm的 ZnGa2O4纳米立方块相互交叠,紧密分布于衬底表面。在氙灯辐照下,ZnGa2O4立方纳米结构光催化降解有机染料主要由光敏化氧化过程实现,染料分子自身的结构、性质以及反应体系的氧气含量是决定光催化活性的关键因素。光催化实验结果显示样品对MB、RhB、CR、MO 4种染料各表现出不同的吸附能力和光催化活性。由于H2O2可作为电子捕捉剂和供氧剂,相比于MOH2O2,MOboth中H2O2的存在使得样品对染料具有持续较高的光催化活性。
猜你喜欢
汽车工程师(2021年12期)2022-01-18
古今农业(2021年2期)2021-08-14
中国交通信息化(2021年1期)2021-06-11
陶瓷学报(2019年6期)2019-10-27
小溪流(画刊)(2017年3期)2017-03-23
中国科技信息(2016年6期)2016-08-31
浙江农业科学(2016年11期)2016-05-04
合成化学(2015年10期)2016-01-17
中国科技信息(2015年24期)2015-11-07
中国科技信息(2015年23期)2015-11-07
