
miR-522-3p通过靶向结合PTEN促进心肌肥厚*
2019-05-08 08:49鲍海龙刘兴德
贵州医科大学学报 2019年4期
张 营, 吴 晟, 鲍海龙, 刘兴德,2***
(1.贵州医科大学附院 心内科, 贵州 贵阳 550004; 2.贵州中医药大学, 贵州 贵阳 550025)
心肌肥厚是心肌细胞为了弥补外来或内在的刺激造成的心脏功能障碍而出现的自主代偿过程,包括心肌细胞体积增大、心肌蛋白合成增加以及肌节目增加[1-2]。加上炎症反应、心肌纤维化改变继而可出现心脏收缩和舒张功能障碍,导致全世界发病率和死亡率最高的心力衰竭疾病的发生[3-4]。微小RNA(miRNA)是一类长约18~22个碱基的非编码小RNA,可结合在靶基因mRNA的3端非编码区(3′UTR)继而调控基因的转录[5-6]。多种miRNA被证实在心血管疾病中发挥重要作用,比如miR-21被证实可调控心肌纤维化中的MAPK通路继而导致心力衰竭加重[7];miR-24可抑制小鼠从代偿性心肌肥厚向失代偿性心肌肥厚的转变过程[8]。本实验旨在证实心力衰竭病理生理过程中miR-522-3p表达出现改变,且miR-522-3p可以参与心肌肥厚的发展过程中。
1 材料与方法
1.1 主要材料
H9C2细胞系由华中科技大学附属同济医院高血压研究所馈赠,miR-522-3p 逆转录引物、miR-522-3p mimic购自广州锐博生物有限公司,Realtime-PCR相关SYBR试剂购自Takara公司,引物合成由汉天一辉远公司完成,pMIR 荧光报告基因载体购自武汉天一辉远公司,转染试剂Lipofectamine2000购自invitrogen公司,双荧光素酶报告检测试剂购自美国Promega公司,AngII购自美国sigma公司,BNP 和ANP 抗体购自美国 Abcam 公司,PTEN、 p-AKT、 total AKT购自美国Proteintech公司。
1.2 方法
1.2.1生物信息学分析 登录NCBI GEO数据库(www.ncbi.nlm.nih.gov/geo)选用芯片分析数据(GSE61741)。
1.2.2动物实验 将8周左右的雄性C57小鼠分为sham组(n=6)和TAC组(n=6),两组适应性喂养2周后行手术。Sham组仅行手术切开暴露胸骨,TAC组暴露胸骨后用7.0手术线进行主动脉结扎。手术后4周进行小鼠心脏超声检测,然后处死小鼠,取出心肌组织以待后续实验。所有动物操作均经过了伦理委员会审查与批准。
1.2.3荧光报告基因实验 将HEK293T细胞接种在24孔板中,待细胞密度至1×106细胞/孔时,将400 ng pMIR-PTEN 3′UTR-wild type与miR-522-3p mimic(100 mmol/L)或mimic control(100 nmol/L)共转染,另将400 ng pMIR-PTEN 3′UTR-mutant载体与miR-522-3p mimic(100 mmol/L)或mimic control(100 nmol/L)共转染,共计4组(即pMIR-PTEN 3′UTR-WT+mimic control组、pMIR-PTEN 3′UTR-WT+miR-522-3p mimic组、pMIR-PTEN 3′UTR-MU+mimic control组、pMIR-PTEN 3′UTR-MU+miR-522-3p mimic组)。细胞培养于37 ℃、5% CO2的培养箱中,24 h后换液,48 h后用冷磷酸盐缓冲盐水(PBS)冲洗细胞,用被动裂解缓冲液裂解细胞(Promega,WI,USA),反复冻融3遍保证细胞充分裂解。使用荧光素酶活性发光计(SIRIUS,Pforzheim,Germany)测量荧光素酶的表达水平。每组选取4个副孔,重复3次独立实验。
1.2.4过表达miR-522-3p实验 将H9C2细胞接种与细胞培养皿中,待密度达70%时,分别将miR-522-3p mimic(100 nmol/L)、mimic control(100 nmol/L)转染进细胞,在37 ℃、5% CO2培养箱中培养,24 h后换液,48 h后收取细胞,提取蛋白及RNA以待后续实验。部分实验中,转染24 h后加用1 mmol/L AngII干预,干预24 h再收取细胞提取蛋白及RNA。
1.2.5实时定量聚合酶连反应(Real-time PCR) 按Trizol法提取sham组与TAC组小鼠的心肌组织,同时提取干预后的H9C2细胞总RNA。测定浓度,选择同样量的RNA(500 ng)进行逆转录。将cDNA与引物、SYBR混合,制备10 μL体系样品, 按95 ℃ 30 s,95 ℃ 15 s、60 ℃ 60 s(40个循坏),95 ℃ 15 s、60 ℃ 15 s、95 ℃ 15 s,完成反应。引物序列见表1。
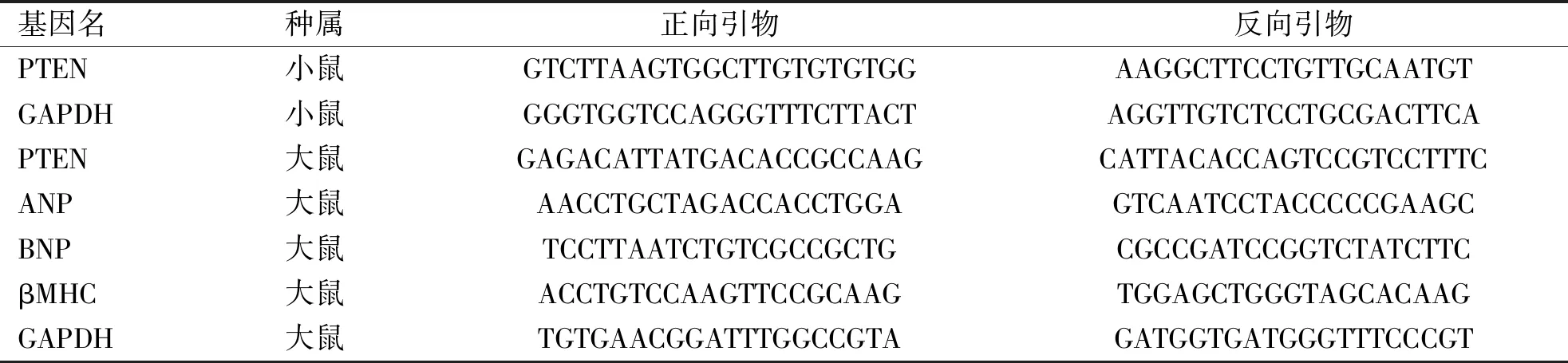
表1 引物序列Tab.1 The list of primer sequences
1.2.6蛋白检测(Western Blot) 按标准法提取蛋白。将蛋白样品与加样缓冲液混合,密封放入沸腾的水浴锅中煮沸5 min,-20 ℃保存。完成相关准备,制胶、上样、电泳、转膜、封闭2 h、一抗孵育4 ℃过夜,二抗室温孵育1 h,曝光。
1.3 统计学分析
采用SPSS 19.0对数据进行分析,计量资料采用均数±标准误表示。同组数据两两比较选用t检验,组内数据采用单因素方差分析,以P<0.05为差异具有统计学意义。
2 结果
2.1 生物信息学分析结果
NCBI数据库中选取芯片(GSE61741)分析疾病组与对照组外周血中miRNA的表达差异,结果显示,与对照组比较,缺血性心力衰竭组和非缺血性心力衰竭组中miR-522-3p的表达均显著升高(P<0.001),见图1A。
2.2 小鼠心肌中miR-522-3p与PTEN表达
对小鼠进行TAC建模,4周后行超声检查,可见TAC组小鼠心肌明显肥厚,射血分数下降(图1B)。实时定量结果显示,TAC组小鼠心肌组织中miR-522-3p表达显著升高[(112.1±20.9)%,P<0.01)],见图1C;同时发现,TAC组小鼠心肌组织PTEN基因mRNA表达明显下降[(-40.0±0.9)%,P<0.01)],miR-522-3p的表达与PTEN表达负相关(r=0.545,P<0.01),见图1。
2.3 miR-522-3p靶向调控PTEN通路
选用Targetscan7.0(www.targetscan.org)对miR-522-3p进行靶向结合位点预测,发现miR-522-3p可结合在PTEN基因3’UTR区,如图2A。荧光报告基因实验结果显示,与mimic control组比较,miR-522-3p mimic明显抑制PTEN 的转录活性(P<0.001),亦抑制了PTEN的表达(P<0.01)。突变结合位点后,miR-522-3p mimic并未影响PTEN的表达活性(图2B);同样的AKT、pAKT,mTOR的表达也受到影响(P<0.01),见图2C。
2.4 miR-522-3p对AngII干预的心肌细胞的影响
AngII干预H9C2细胞后,可见PTEN蛋白和mRNA表达水平下降,miR-522-3p使PTEN的表达下降得更明显,差异有统计学意义(P<0.01)。心肌肥厚相关分子指标ANP、BNP、βMHC表达上升,而miR-522-3p mimic使这种上升更加明显,差异有统计学意义(P<0.05),见图3。
3 讨论
心力衰竭是最常见的心血管疾病且是大部分心血管疾病的终末阶段[9],而它的生物学基础--心肌肥厚可以受多种信号通道调控,如Akt、PI3K、MAPK、AMPK、GPCRs等信号通路[1]。研究者认为这些信号通路的调控者可以调控整个心肌肥厚过程达到改善或逆转心肌肥厚的目的,继而治疗心力衰竭,比如PINK1基因可以调控AMPK的表达影响心肌细胞自噬过程继而影响心力衰竭[10]。故而GRCPs下游β受体阻滞剂被应用于临床心力衰竭患者,CIBIS-II、MERIT-HF、COPERNICUS三大临床实验证实β受体阻滞剂的应用大大改善了心力衰竭患者的预后[11-13]。
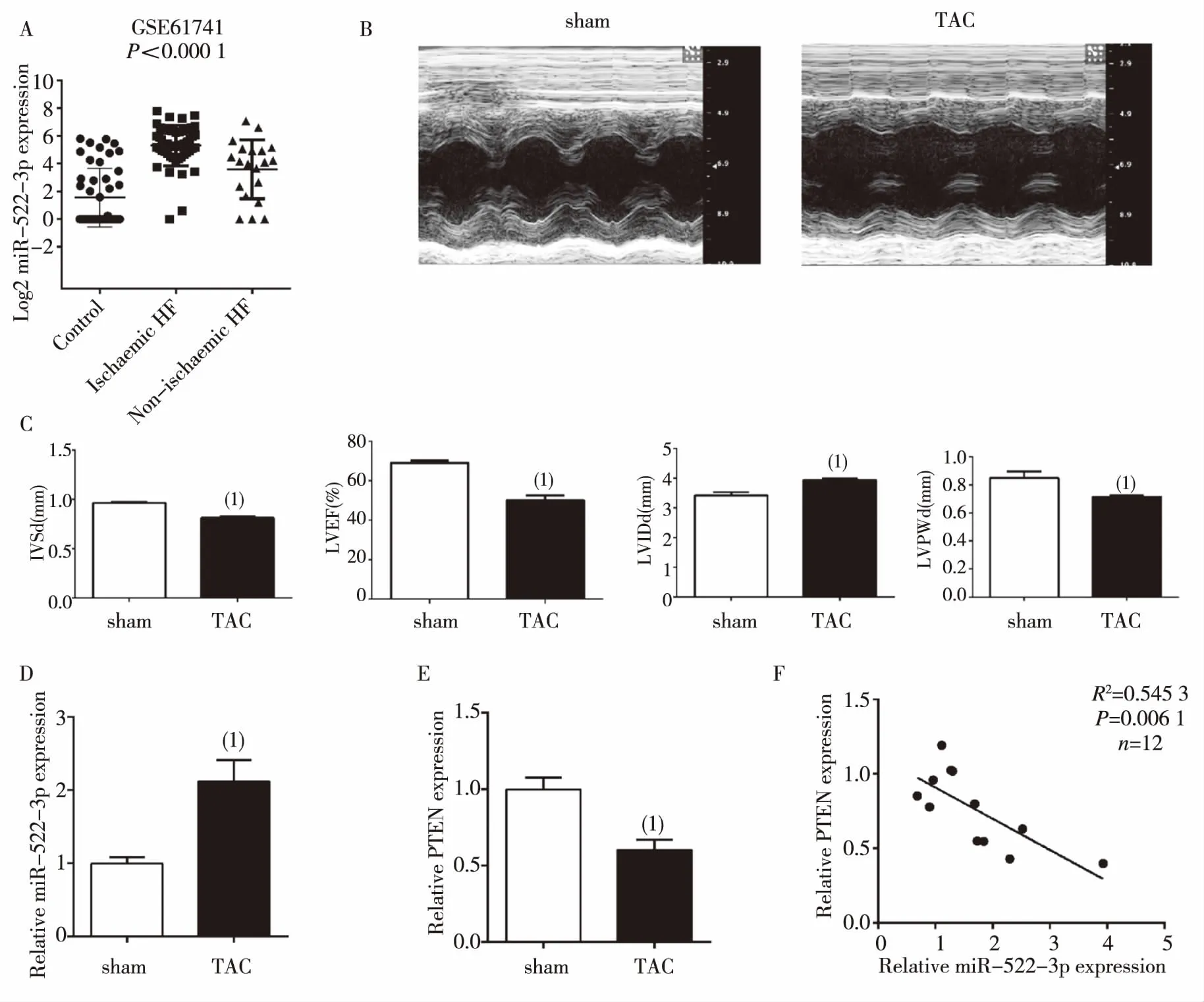
注:A为GSE芯片数据,B为心脏超声结果,C为心功能指标,D为中心肌组织miR-522-3p的表达,E为PTEN表达,F为miR-522-3p与PTEN表达的关联性分析;(1)与对照组比较, P<0.05图1 心力衰竭与对照组中miR-522-3p的表达Fig.1 The expression of miR-522-3p in heart failure group and control group
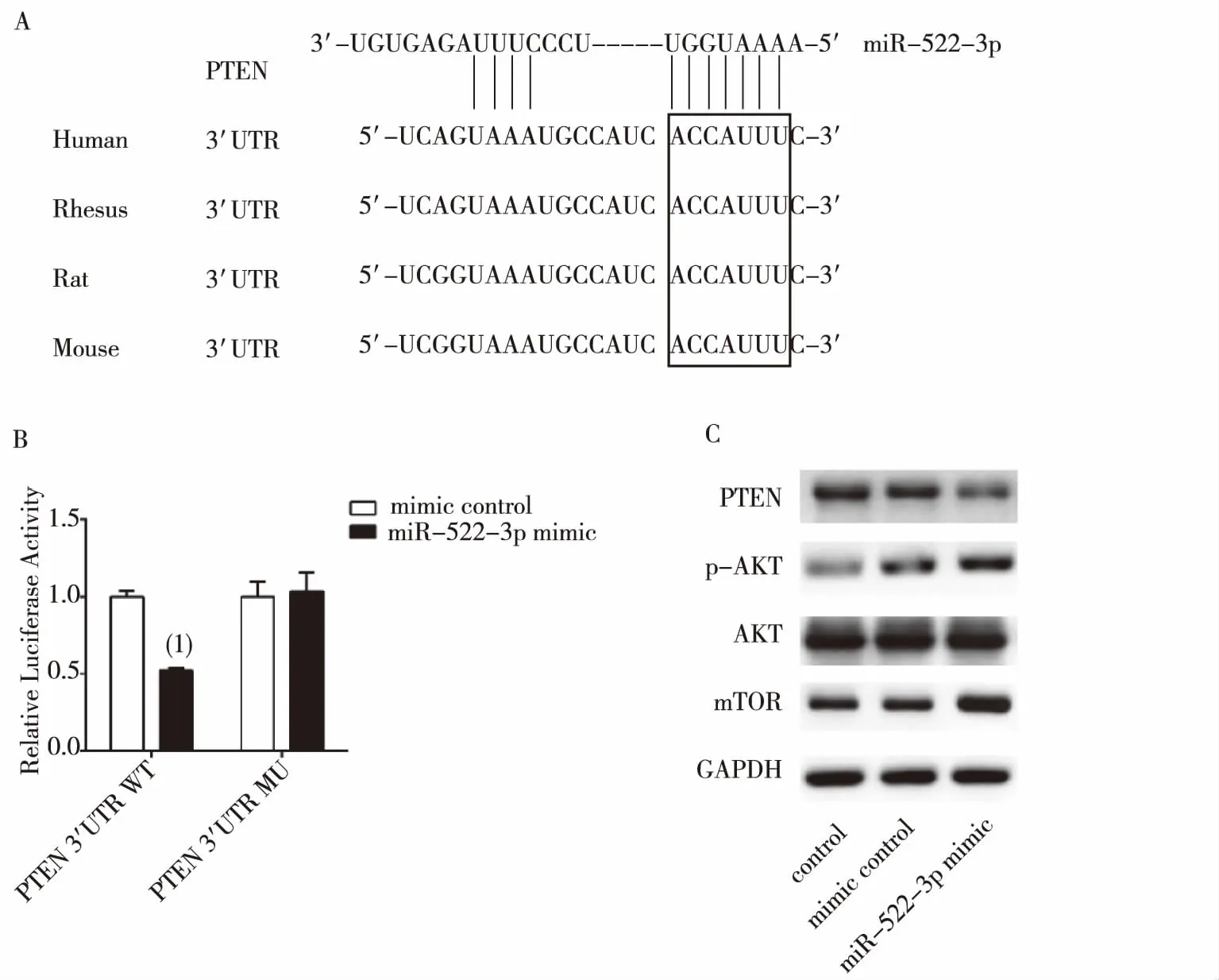
注:A为预测miR-522-3p结合在PTEN基因3′UTR区,B为在293T细胞系中荧光报告基因检测miR-522-3p与PTEN的相互作用,C为miR-522-3p调控PTEN/Akt/mOTR通路;(1)与对照组比较,P<0.001图2 miR-522-3p靶向调控PTEN通路情况Fig.2 miR-522-3p regulated PTEN/Akt/mTOR pathway through targeting PTEN
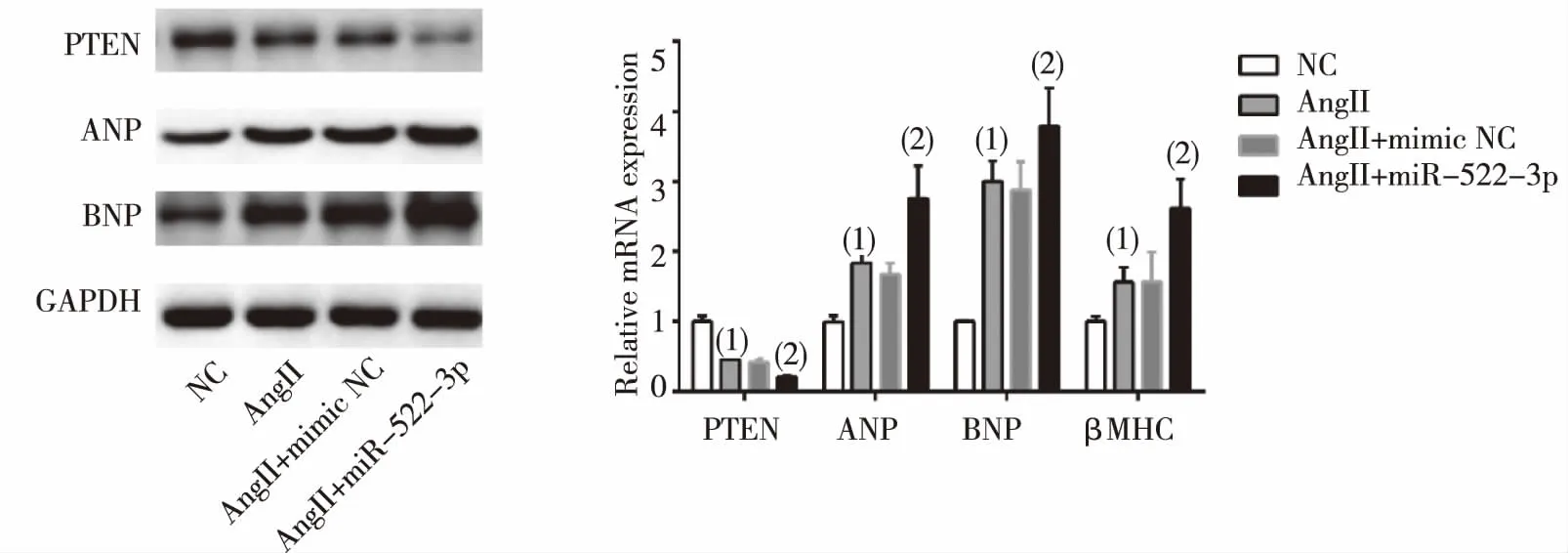
(1)与对照组比较,P<0.05;(2)与AngII+mimi NC组比较,P<0.05图3 miR-522-3p对AngII干预的心肌细胞的影响Fig.3 The effect of miR-522-3p on Ang II-mediated Cardiomyocytes
但随着心力衰竭的发病率致死率逐年增高[14],人们不满足于仅仅使用β受体阻滞剂治疗心力衰竭,更多心肌肥厚过程中的调控者被人们关注起来。脑啡肽酶抑制剂(ARNI)被证实可以产生良好的心力衰竭治疗作用[15],同时miRNA也是一大热点。miR-522-3p最早被报道于肥胖相关疾病中[16],结合在肥胖高度相关的基因上,结合处SNP位点突变破坏结合影响基因的表达,继而随着SNP突变出现不同肥胖表型。后续多个研究报道显示miR-522-3p可以促进癌细胞的增值、迁移且与肿瘤的预后相关[17-19]。本研究结合GSE数据库,首次发现在心力衰竭患者组中循环miR-522-3p的表达明显高于对照组,但血中循环miRNA的表达与组织miRNA表达是具有差异性的。为了验证心力衰竭患者的心肌组织中miR-522-3p也是高表达,本实验设计了小鼠TAC模型验证。造模后4周心脏超声显示TAC组小鼠心肌明显肥厚且心功能下降,提示造模成功。对心肌组织进行实时定量PCR测定,发现miR-522-3p的表达明显升高。由此看来,miR-522-3p可能不仅仅参与肿瘤相关疾病。
与此同时,该研究还发现心力衰竭小鼠中PTEN的表达下降,与miR-522-3p的表达负相关。通过靶点预测,发现miR-522-3p靶向结合在PTEN基因的3’UTR区,这与大多miRNA-mRNA结合方式吻合。而PTEN被发现在心肌细胞、成纤维细胞和内皮细胞中普遍表达,且可负性调控PI3K信号通路[20]。而且PTEN/Akt信号通路,近来亦被证实是与心肌肥厚密切相关的[21]。本研究证实,过表达miR-522-3p后PTEN的表达活性明显下降,且PTEN/Akt/mTOR信号通路的表达受到明显影响。这个发现为miR-522-3p调控心肌肥厚提供了理论支持点。为了证实这种可能,本实验使用了体外心肌肥厚细胞模型。在AngII诱导H92C细胞中,ANP、BNP、βMHC这些心肌肥厚相关分子指标明显上升。过表达miR-522-3p,这些指标进一步上升,提示心肌肥厚加重。本研究认为,miR-522-3p通过调控PTEN/Akt/mTOR信号通路继而调控心肌肥厚。
综上所述,本实验首次发现心力衰竭中miR-522-3p表达上升。PTEN基因表达下降,二者呈负性相关。继而在H9C2细胞中过表达miR-522-3p,发现明显抑制PTEN/AKT信号通路。与此同时,在AngII干预的H9C2细胞模型中,过表达miR-522-3p,增加心肌肥厚相关的ANP、BNP、βMHC蛋白的表达。但本实验没有在动物层面进行进一步验证miR-522-3p是否会对心脏超声影像学、心肌组织形态学等层面造成影响,缺乏更多更严谨的证据证实本研究的设想。不过就目前结果,为后续实验提供了一个新的思路,也为抗心肌肥厚治疗提供了一个新靶标。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
作文教学研究(2016年1期)2016-07-05
海南医学(2016年8期)2016-06-08
中国病理生理杂志(2015年8期)2015-12-21
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年3期)2015-06-10
