
工业源挥发性有机物治理功能材料研究进展
2019-04-29 10:48:04王晨昊颜炳君王继荣牛何晶英高国瑜陈浩程伶俐吴明红焦正
自然杂志 2019年2期
王晨昊,颜炳君,王继荣,牛何晶英,高国瑜,陈浩,程伶俐,吴明红†,焦正††
①中船第九设计研究院工程有限公司,上海 200063;②上海大学 有机复合污染控制工程教育部重点实验室,上海 200444
挥发性有机物(volatile organic compounds,VOCs)是在标准大气压101.3 kPa下,初馏点小于等于250 ℃的所有有机化合物的总称,主要包括烃类、卤代烃、醛类、酮类、芳香烃以及多环芳香烃等,其中以苯系物最多,约占20 %[1]。VOCs来源广泛,可分为自然源和人为源。自然源包括植被排放、野生动物排放、森林火灾、沼泽的厌氧过程等,其中以植物排放为主。人为源比较复杂,包括机动车末端排放、油气挥发、有机溶剂使用、各种工艺过程、石油冶炼、垃圾填埋等,主要来源于工业。当该类物质存在于大气中时,即使其浓度很低,也会对环境以及人类的健康构成直接的威胁[2-3]。对于我国频频爆发的雾霾天气,VOCs是雾霾形成的重要前驱物,扮演着重要的角色。此外,VOCs还是形成光化学烟雾的重要前驱体[4-5]。因此近些年来,VOCs的治理越来越受到人们的重视。2013年中国国务院印发的《大气污染防治行动计划》要求“从生产、运输、使用等环节对VOCs进行排放治理”,这意味着VOCs已经成为我国大气污染防治的重点。2017年,我国颁布的《“十三五”挥发性有机物污染防治工作方案》,计划到2020年,实现在重点地区、重点行业VOCs污染排放量总量下降10 %。可见,面对日益严格的VOCs控制的法律法规,产业界对VOCs控制技术的需求十分迫切。
目前,针对VOCs的特点,常用的治理技术有吸附法[6-8]、光催化法[9-12]、催化燃烧(氧化)法[1,5,13-16]以及生物过滤法[17]等。实现这些技术所使用的新型功能材料也就随之成为了VOCs治理过程的核心,功能材料的优劣将直接影响VOCs治理的效果。本文重点综述了近年来VOCs治理关键功能材料的研究进展,包括影响VOCs吸附剂和催化剂性能的主要影响因素,以及各类功能材料改进的方法与手段,希望能够为将来VOCs治理材料的设计与研发提供一定的参考。
1 VOCs吸附材料
吸附法是利用吸附剂将气相的VOCs吸附到固相吸附剂表面,从而使气体得到净化的方法。一般认为,吸附法是处理低浓度VOCs最经济、绿色的方法[3]。常用的吸附剂主要是一些具有大比表面积或多孔的材料,如以活性炭(AC)和石墨烯为代表的碳基材料、沸石分子筛及金属有机骨架类(MOF)无机吸附材料、高分子有机材料以及它们的复合材料等。对于VOCs吸附材料而言,其性能以及工业应用的价值主要取决于吸附容量、扩散和传质效率以及可重用性[7]。
1.1 碳基吸附材料
碳基吸附材料是最为常见的一类吸附剂,有着很高的比表面积以及孔隙率,对如芳香族环烃类的大分子有机物有着极强的吸附能力,同时碳基吸附材料的造价较其他类型吸附材料而言相对较低,吸附剂再生也较为方便,因此被广泛地应用在气体吸附领域[7]。但是,传统的碳基吸附材料也存在着一些难以克服的缺陷。以AC为例,不仅AC自身的湿热稳定性较差,而且使用传统方法制备的AC其孔径分布一般在微孔(<2 nm)和大孔(>50 nm)范围内,这直接导致传统AC对小分子量VOCs的去除效率不高[7,18]。目前对新型碳基吸附材料的研究主要围绕着碳基吸附材料吸附VOCs的机理、吸附模型以及新型碳基吸附材料的研发。
Lillo等[19]在研究AC对甲苯以及苯的吸附行为后指出,AC对上述两种物质的吸附量受微孔控制,AC中微孔的孔容越大对应的气体吸附量也越大。宋剑飞[20]在AC构效与VOCs吸附性能的关系方面做了大量的基础性研究,认为良好的孔径分布有利于提高吸附气体在AC内部的扩散,且孔径直径为吸附质分子动力学直径2~3倍的微孔吸附性能最佳。对AC进行表面改性以提高AC吸附量并实现选择性吸附也是研究的热点问题。一般情况下,AC表面含氧官能团中酸性化合物含量越高,AC对极性化合物的吸附效率越高;反之,则对非极性或极性较弱的物质吸附越强[21-22]。Mohammed等[23]使用KOH在微波的作用下活化椰壳炭得到PHAC,并在此基础上又进一步使用氨处理,以氮官能团部分置换PHAC表面的含氧官能团,从而增强PHAC的疏水性质得到PHAC-AM,并研究了二者对苯以及甲苯的吸附能力。实验表明:氨处理的PHAC-AM对苯、甲苯的去除率分别为91 %和92.3 %,相比处理前提高了近10 %,并且PHAC-AM对苯以及甲苯的吸附符合Langmuir吸附等温模型。
目前使用新型碳源制备AC也是研究的方向之一。 Zhu等[24]首先使用聚多巴胺合成的球形模板在800 ℃下碳化,随后按质量比1:4以及1:6与KOH混合(分别标记为MGBC4和MGBC6),在N2保护下以700 ℃进行活化。通过XRD、拉曼光谱以及XPS手段证明了MGBC均含有石墨化的碳,且MGBC4的石墨化程度大于MGBC6——这可能是因为MGBC6较MGBC4有着更多的微孔结构,而这些微孔破坏了样品的石墨化程度。在随后进行的对甲苯以及苯的吸附实验中,MGBC在低压区(P/P0<0.02)表现出了比MIL-101以及 HKUST-1更优的吸附性能。文章认为,由于MGBC样品中存在部分石墨化的结构,使得吸附剂可以和苯系物形成强烈的π-π作用;此外这种结构使得吸附剂较一般MOF材料表现出更强的疏水性,有利于在湿润条件下吸附剂对苯系物的选择吸附。Qian等[25]则以偏二氯乙烯以及苯乙烯的共聚物为原料制备得到了活性炭微球ACM,BET测试表明该材料有较大的比表面积1 421 m2/g,且主要均由微孔提供(94.4 %)。对该材料吸附氯甲烷(CH2Cl2、CHCl3、CCl4)以及碘甲烷(CH3I)的性能进行了研究,结果表明ACM对上述物质的吸附能力均强于市场上常见的使用椰壳制备的AC。Wang的团队[26]使用EISA路线合成了具有较大比表面积(1 762 m2/g)的有序介孔碳(OMC),探讨了其对苯、环己烷以及己烷的吸附行为。由于该材料的孔径呈双峰分布且主要集中在5.0 nm以及1.8 nm,较选取的VOCs气体分子大,故OMC的总孔容均可以得到充分利用,在298 K下该材料对苯、环己烷和己烷的吸附量分别达到了17.34、14.19和11.97 mmol/g。此外,这种分峰分布的介孔结构使OMC较常用的碳材料(如AC、碳分子筛)有更大的努森扩散率。废旧的碳材料也被用于制备活性炭材料。László等[27]使用废旧的PAN以及PET为碳源,并使用水蒸气在900 ℃下活化制备AC;Kartel等[28]与Bratek等[29]则利用废弃的PET瓶为碳源制备了活性炭。近期Choma等[30]利用粉碎后的废旧CDs以及DVDs为碳源在500 ℃下进行碳化,随后分别使用KOH以及CO2在不同实验条件下对样品进行活化,并研究了其对CO2、H2以及苯的吸附性能。其中样品C-KOH-4(KOH与C的质量比值为4)和C-CO2-8(CO2活化时间为8 h)表现出了对苯最佳的吸附容量,分别为13.6和15.4 mmol/g。
碳纳米管(CNT)也是一类常见的碳基材料,近年来对其吸附VOCs的行为做了相当多的研究。Li等[11]使用反相气相色谱法研究了15种VOCs气体在不同相对湿度下(RH)在多壁碳纳米管上的气/固分配系数,并通过线性溶剂自由能关系法(LSER)建立了VOCs与MWCNT吸附作用的关系。通过LSER分析,Li认为VOCs在MWCNT表面的吸附机理主要是基于π/n-电子对之间的相互作用以及氢键酸度,即VOCs在MWCNT表面的吸附容量主要由MWCNT自身的石墨结构以及表面的含氧官能团提供。随着RH的提高,由于MWCNT表面被水分子占据,VOCs吸附容量大幅下降。LSER回归分析表明,主要的吸附机制为空腔形成、偶极性/极化率和氢键碱度。类似地,Cheng等[31]通过理论线性溶剂化能关系(TLSER)认为,VOCs在MWCNT表面的吸附主要是因为范德华力和氢键的相互作用。
碳化物衍生碳(CDC)是一种新型多孔碳材料,其通过选择性地蚀刻碳化物中的非碳原子制备而成[32-33]。由于CDC材料巨大的比表面积和孔隙容积以及可调控的孔径分布,越来越受到学界的重视。目前CDC材料主要被应用于气体储存与吸附[34]、电极材料[35]、超级电容器[36]及催化剂载体[37]等。对于气体储存而言,目前的研究主要集中在储存氢气[34,38-39]以及甲烷[40]上;作为气体吸附剂来说,CDC近年来的研究重点是对CO2的吸附[41-42],对VOCs的研究则相对较少。Wang等[18]首次报道了使用氯气蚀刻碳化钛(TiC)法制备的CDC材料对小分子VOCs的吸附性能,研究表明:CDC较以木炭或椰子壳制备的AC表现出对小分子VOCs更强的吸附能力。文中把这一现象主要归因于CDC具有更大的比表面积和微孔孔容,以及窄的孔径分布(0.7~1.5 nm)。同时,该CDC材料的再生过程与AC相似,在110~150 ℃下即可完成。
作为新型碳基材料的代表,氧化石墨烯膜(GOM)具有严格的二维结构、超高的比表面积,是一种优异的吸附材料。与具有固定尺寸的其他碳基材料不同,GOM可以通过精确调控层间距,对吸附或过滤对象实现选择性筛分和净化。
目前GOM层间距的调控,可通过插入大的纳米材料以及交联大分子和刚性分子加宽层间距,或者通过还原GOM减小其层间距[43-44]。吴明红课题组[45]通过利用不同的水合离子调控层间距,实验表明:不同的阳离子(K+、Na+、Ca2+、Li+和Mg2+)本身可以决定和控制小至纳米尺寸的GOM层间距,并且实现了GOM层间距1/10 nm的精确控制(图1)。 通过密度泛函理论计算表明:其他阳离子(Fe2+、Cu2+、Cd2+、Cr2+和Pb2+)与石墨烯具有更强的阳离子-π相互作用。这表明其他阳离子可以用来产生更宽的层间距[46]。
尽管石墨烯的研究历史很短,但已经展现了众多的潜在应用。在VOCs的排放治理方面,由于VOCs具有复合污染的特点,通过调控不同层间距的石墨烯,对各VOCs组分进行选择性吸附,可有效分离和吸附经济效应高的VOCs组分。
1.2 无机吸附材料
目前无机吸附材料主要以沸石分子筛和MOF类材料为主。沸石分子筛是一类典型的多孔物质,具有规整的微孔孔道结构。自20世纪Richard M. Barrer成功合成出低硅铝比的沸石分子筛以来,它就被广泛地用作为吸附材料、离子交换材料以及酸催化材料[47-48]。近年来,沸石分子筛由于其优异的性能也被逐渐应用到VOCs治理领域,但是大部分沸石分子筛只具有单一的微孔孔道结构,扩散阻力较大。同时,沸石分子筛较窄的微孔结构也使其仅能吸附分子直径较小的分子,而且与其他纳米材料一样,沸石纳米颗粒由于本身巨大的比表面能很容易造成颗粒间的团聚,造成有效表面积的大幅下降[7]。因此,近些年的研究主要集中在对沸石类材料的孔径进行改进以及防止沸石材料的团聚上。
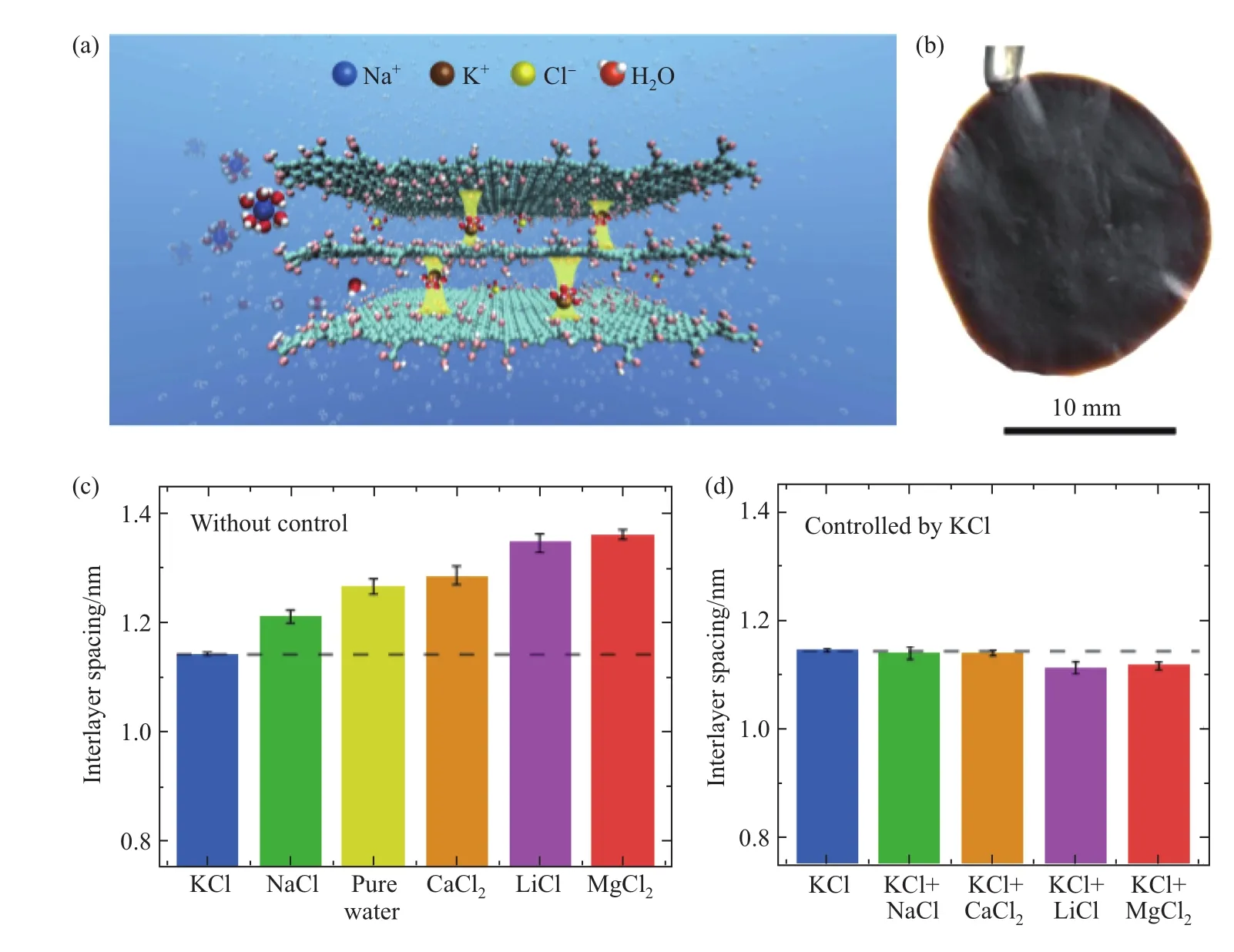
图1 无支撑阳离子调控的GOM的层间距:(a)在GOM中K+如何确定和固定层间距的示意图,其他阳离子被排斥,而纯水可以通过(黄色柱表示水合K+对层间距的固定作用);(b)由5 mg/mL的GOM悬浮液滴注制备的无支撑GOM; (c)GOM在纯水或不同的0.25 mol/L盐溶液中的层间距;(d)先在KCl溶液中浸泡,再浸入各种盐溶液中的GOM的层间距[45]
将纳米的沸石颗粒负载到有大孔结构的基底上是解决颗粒团聚的常用方法之一,同时该方法还可以形成新的多级孔结构,有利于降低气体分子的渗透阻力从而提高客体分子的传质速率[7]。Yuan等[7]通过静电自组装的方法,在聚二烯丙基二甲基氯化铵(PDDA)改性硅藻土的表面原位负载上纳米Silicalite-1颗粒(M-Dt/Sil-1nano)。在PDDA改性前,硅藻土表面带有的负电荷与Silicalite-1表面的负电荷形成静电排斥作用,故在未改性直接负载的样品Dt/Sil-1nano表面负载的Silicalite-1较少(负载率32.1 %)且不均匀(图2(a))。在PDDA改性后,硅藻土表面的电荷转变为正电荷,与Silicalite-1形成吸引作用,故M-Dt/Sil-1nano表面的Silicalite-1分布均匀且负载率达到60.2%(图2(b))。在苯的吸附实验中,M-Dt/Sil-1nano对苯的动态吸附量达到246.0 mg/g。

图2 SEM图像[7]:(a) Dt/Sil-1nano;(b) M-Dt/Sil-1nano
MOF是一种新兴的混合多孔材料,由于其具有高比表面积、高催化活性、优良的热/化学稳定性、可调的孔径和表面化学官能团,以及对气体优异的吸附性能,被认为是VOCs优异的吸附剂材料。但是,MOF开放的金属位点以及相对较弱的金属-氧化物配位键,使其极易受水分子的影响而导致自身结构的破溃[8,49-51]。近年来,对MOF材料作为VOCs吸附材料的研究主要集中在提高MOF结构稳定性,对MOF材料孔道及表面微环境的功能化设计以及一些吸附机理的基础性研究上。
Xian等[3]对水蒸气、1,2-二氯乙烷(DCE)、乙酸乙酯(EA)以及苯分子在MIL-101(Cr)表面的竞争吸附做了细致的研究。程序升温脱附实验(TPD)表明:水蒸气、DCE、EA和苯的脱附活化能大小依次是水蒸气(=72.9 kJ·mol-1)>DCE(=47.14 kJ·mol-1)> EA(=41.9 kJ·mol-1)> 苯(=38.16 kJ·mol-1)。这表明水分子与MIL-101表面的相互作用强于所选取的VOCs分子,水蒸气的存在将严重影响MIL-101对VOCs的吸附。因此,MOF材料作为VOCs的吸附剂时,应对其表面进行疏水性修饰以减少水分子对吸附效果的影响。Vellingiri等[8]对VOCs特别是SVOCs(半挥发性有机物)在典型MOF材料(MOF-5、Eu-MOF以及MOF-199)上的吸附行为做了大量基础性研究,补充了以C2~C5、挥发性脂肪酸 (VFAs)、苯酚为代表的SVOCs的吸附参数。Vellingiri的实验以及DFT模拟还进一步证明了MOF-199对VOCs的吸附主要基于MOF材料框架中存在的开放金属位。Gutierrez的科研团队[52]利用反相气相色谱法探讨了同网状金属有机骨架(IRMOF)空腔大小与化学环境对VOCs吸附性能的影响。实验中Gutierrez采用刚性的二元羧酸链接Zn4O四核簇以形成立方框架,并在3D方向进行生长,最终形成正方形通道。为了探索MOF材料空腔大小对VOCs吸附性能的影响,实验中分别选取对苯二甲酸、2,6-萘二甲酸以及4,4'-联苯二甲酸作为连接基团制备了同网状金属有机骨架并依次标记为IRMOF-1、IRMOF-8和IRMOF-10。实验表明:对于正构烷烃而言,吸附量和吸附强度与被吸附物的大小有关;而对于极性化合物来说,吸附量和吸附强度与π-电子丰富区域有关。
Yang的团队[51]首次在溶液中通过咔唑桥接含有正丁基的有机配体L与Cu(NO3)2的反应,成功合成出非贯穿2D Cu(Ⅱ)-MOF材料(图3(a))。该2D材料通过以-ABAB-的方式在c轴方向堆叠可以形成类方形的孔道(图3(b)),同时由于配体L上所有的正丁基均面向孔道中心,故形成的方形孔道是疏水的。该材料可以实现对VOCs(如CH2Cl2/CHCl3)以及BTEX(苯、甲苯、乙苯和二甲苯)的可逆吸附。此外,该MOF材料能够在液相中对VOCs以及BTEX进行有效分离。对于含氯烃而言,这种选择吸附性主要源于分子极性;而对BTEX而言,则主要源于疏水作用。
1.3 复合吸附材料
由于上述的各类材料在实际应用中各有优缺点,为了更符合VOCs治理的实际情况,许多科研团队将重点放在复合材料的研发设计上,希望通过不同材料间的协同作用,达到取长补短的目的。石墨烯类材料由于具有独特的层状结构、巨大的比表面积以及改性后表面丰富的官能团,近年来被广泛地应用到VOCs吸附材料的复合改性上。
Zhou的团队[6]通过将沸石咪唑酯骨架结构材料(ZIFs)与氧化石墨烯(GO)复合并调控GO的含量(0~15 %),实现对复合吸附剂表面形貌的调控。在对CH2Cl2吸附的实验中,所有复合材料均表现出较单一ZIFs更优越的吸附性能,且当GO的含量达到15 %时复合材料的吸附性能最优,达到240 mg/g。文章中将这一现象归功于ZIFs与GO间强烈的协同作用以及GO所引入的大量表面官能团对CH2Cl2分子的强烈相互作用。
Li等[49]将MOF材料Cu-BTC和GO在无溶剂的条件下通过机械化学合成法进行复合,最终产物Cu-BTC@GO在298 K下表现出对甲苯优异的吸附性能,吸附量达到838.5 mg/g,较未复合的Cu-BTC提高了47 %。此外,Cu-BTC@GO表现出对水极强的稳定性,在水中浸泡10 h其形貌依旧保持完好(图4),比表面积也依旧保持有1 205 m2/g,较浸泡前1 362.7 m2/g略有下降。然而,未与GO复合的Cu-BTC在浸泡后则发生了严重的形貌垮塌,比表面积也从1 188.3 m2/g下降至 20.7 m2/g。实验表明,将MOF材料与GO复合可有效提高其水稳定性。

图4 SEM图像:(a)Cu-BTC;(b)Cu-BTC-10h;(c)Cu-BTC@GO;(d)Cu-BTC@GO-10h[49]
Bai等[53]则是以酚醛树脂和GO为前驱体,通过电纺丝及在氩/氢气氛中碳化的方式,成功制备出还原氧化石墨烯(RGO)/碳复合超细纤维(PCGF-H)(图5(a))。由于RGO的引入,该材料表现出比未复合RGO材料更强的疏水性以及对VOCs更强的吸附能力。水分子与sp2杂化的碳基面的吸附作用主要靠较弱的色散力(3~6 kJ·mol-1),而与sp3杂化碳以及含氧官能团间的作用主要是较强的极性作用(40 kJ·mol-1)。在氩/氢气氛碳化的过程中,GO被H2还原为RGO,表面的含氧官能团大大减少,而PCGF-H表面sp2杂化的碳含量上升。以上两点减弱了水分子与PCGF-H的相互作用,提高了其疏水性。其原理示意图如图5(b)所示。
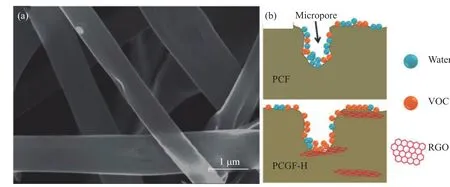
图5 (a)PCGF-H的SEM图像;(b)VOCs与水分子在PCF和PCGF-H表面竞争吸附的示意图
2 VOCs光催化材料
VOCs的光催化降解处理是一种温和、清洁、绿色的高级氧化处理手段。光催化反应的主要过程:①首先半导体价带(VB)中的电子,吸收光子能量跃迁至导带(CB)中形成自由电子,在VB中则留下了空穴;②在半导体自建电场的作用下,电子与空穴向材料表面进行迁移,在迁移的途中电子与空穴可能发生复合而失去活性;③迁移至表面的电子、空穴分别和催化剂表面吸附的电子受体和电子给体发生还原与氧化反应,生成大量的自由基,自由基可以将VOCs分解至矿化[54]。因此,高效的光催化剂应能够高效吸收光能形成光生电子-空穴对,并有效地将e-和h+分离[55-56]。
2.1 TiO2光催化材料
TiO2由于其低廉、安全、稳定、光催化活性高等特点成为目前应用最广泛的光催化材料[9]。但是,禁带宽度过大(金红石相:3.0 eV,锐钛矿相:3.2 eV),只对紫外波段光有响应,以及电子-空穴对复合率高是TiO2材料的严重缺陷[10],因此拓宽TiO2的可见光响应,从而提高光能利用效率,降低电子-空穴对的复合率是TiO2光催化剂主要的科研方向。针对上述两点,到目前为止的研究证明,降低电子-空穴对复合率以提高其向TiO2表面迁移速率的方法,主要有贵金属表面负载[57]、金属阳离子掺杂[10]以及构建半导体异质结[58]等。拓展可见光响应则可以通过非金属杂质原子(如C、N、B、S等)掺杂的方式,利用贵金属纳米粒子表面等离子体共振效应或者与具有可见光响应的窄禁带半导体材料(如CdS、CdSe等)复合得以实现[48]。
对于使用TiO2光催化降解VOCs气体而言,要求光催化剂具有较大的比表面积,为光催化反应提供更多的反应活性位点,并且催化剂材料需要便于回收以符合实际生产应用的需求。将纳米TiO2催化剂材料固定在载体材料表面是常用的手段,该方法不仅可以改善纳米材料的团聚现象,增大催化剂比表面积,还使得催化剂材料更便于回收。目前常用的载体材料主要有硅胶、多孔铝、沸石分子筛、黏土以及碳材料等。
Tasbihi的团队[59]将硅/二氧化钛溶胶作为黏着剂,将等质量混合的商业Degussa P25以及Millennium PC500 TiO2固定在有保护层保护的铝片上,并研究了该催化剂材料中黏着剂以及铝片保护层对其光催化降解甲苯性能的影响(图6)。实验结果表明,含有黏合剂以及保护层的样品titania/binder/Al-p表现出最佳的光催化性能。Tasbihi将其归因于黏合剂提高了TiO2在Al表面的分散度以及均匀度,保护层则在防止催化剂中毒的同时提高了甲苯在催化剂表面的吸附量。
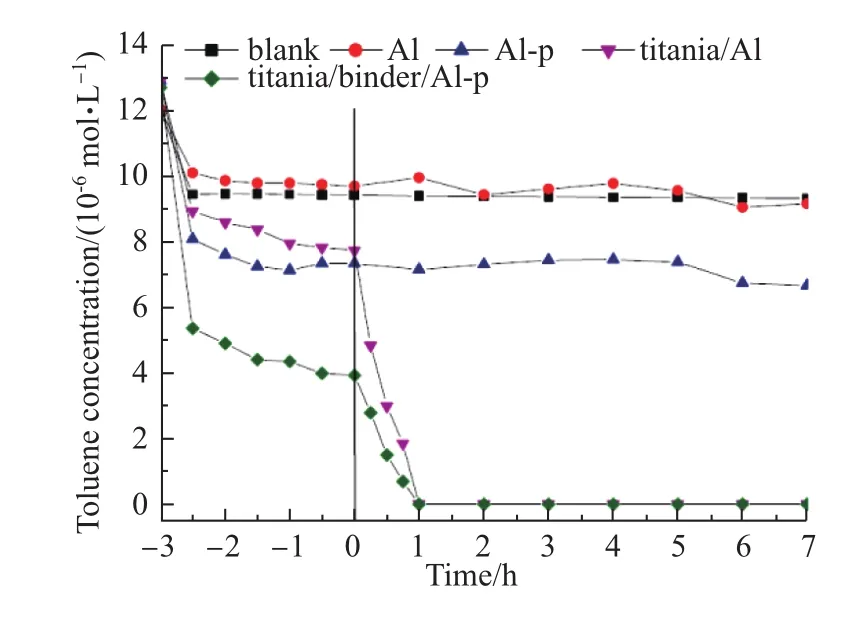
图6 UVA照射前后甲苯浓度变化图[59]
Šuligoj等[9]首次利用钛酸为前驱体在NaOH以及HCl的作用下制备出锐钛矿相纳米TiO2胶体溶液(AS),随后以摩尔比1:1的形式固定在多孔硅SBA-15上形成AS/SBA-15。该材料的TEM图像(图7(a))表明,纳米TiO2的粒径为5~10 nm,并且成功进入了介孔SBA-15的孔道中形成包覆结构,这种结构提高了TiO2的结晶度。为验证其光催化性能,Šuligoj首先将AS/SBA-15通过刷沉积(brush deposition)的方式固定在薄玻璃表面,随后在UVA照射下对甲苯以及甲醛光催化降解。实验结果表明,该材料对甲苯以及甲醛的光降解率分别为100 %和91 %,其中对甲醛的降解效率优于未复合SBA-15的AS以及商用的P25和PC500(AS/SBA-15 > P25 > PC500 > AS)。实验结果见图7(b)。此外,AS在负载SBA-15后其交叉频率(TOF)较负载前提高了近6倍。

图7 (a)光催化材料AS/SBA-15的TEM图像;(b)光催化降解时甲醛的体积分数随时间的变化[9]
碳材料由于其丰富的结构、较高的比表面积、低廉的价格而受到学界广泛的关注。此外,碳材料尤其是石墨碳,其拥有离域共轭的π电子体系,可以将光生电子快速转移,因此在作为载体材料时,不但可以缓解光催化材料团聚,还可以有效降低电子-空穴对的复合率,提高光催化效率[60]。Ouzzine等[61]使用溶胶-凝胶法将TiO2负载到球状AC上,该复合光催化材料TiO2/AC对低浓度的丙烯表现出良好的性能。值得注意的是,该复合材料在UVA段的催化活性优于UVC段的催化活性。
金属铜Cu由于其优良的性质也被用于TiO2的改性。首先,相对于其他贵金属而言,Cu储量较大且价格低廉;其次,单质Cu及其化合物(Cu2O、CuO等)均可以形成电子介体和异质结,从而扩大吸收波长的范围,有效降低电子-空穴对的复合率[62]。Stucchi等[10]利用20 kHz的高能超声(US)将Cu修饰到商业TiO2(Kronos)表面,并探究了Cu含量对甲醛以及乙醛的可见光催化性能。Stucchi在文章中指出,US不能改变TiO2的表面形貌,但可以有效地在其表面形成Cu单质及其氧化物,并且在一定范围内较高的Cu含量能有效提高TiO2的光催化性能(文中实验条件下Cu负载率在质量分数为40 %时催化剂性能最佳),但Cu含量过高,则由于形成太多的活性点位造成电子-空穴复合率上升。Martínez等[63]使用溶胶-凝胶法制备出Cu掺杂TiO2,当Cu的掺杂量至0.2 %(质量分数)时表现出对三氯乙烯(C2HCl3)最佳的光催化性能。实验结果表明,较小的晶粒尺寸、最低的Ti—O—Ti振动能以及最适当的微孔性特征是其具备最优光催化性能的原因。
2.2 其他光催化材料
由于TiO2自身较大的禁带宽度,许多科学家将研究的目光转向其他禁带宽度较小、光生载流子寿命更长的材料上。
Bi2WO6是一种禁带宽度为2.69 eV的混合金属氧化物,因其能被可见光激发而受到重视。Zhang[64-65]以及Yu[66]的团队均使用水热法成功制备出纳米Bi2WO6材料且均表现出优异的可见光响应。Zhang等[60]则首次在未使用模板的情况下制得花状Bi2WO6材料。但是,Bi2WO6也存在一些缺陷,如对光吸收的能力较差以及电子-空穴对复合较快等[12]。Qian等[12]通过湿法浸渍法将碳量子点(CQDs)与水热法制备的Bi2WO6进行复合,利用CQDs可以吸收UV和近可见光以及将光诱导电子转移、储存的特性,降低了电子-空穴对的复合率,提高了其对丙酮和甲苯气体的光催化性能。Ullah的团队[67]在二元金属材料对VOCs的光催化性能方面做了比较多的工作。他们采用溶液顺利制备出Bi2WO6、AgVO3、AgTaO3、BiTaO4、Cd2Ta2O7、 InTaO4、CoTa2O6、CeTaO4以及EuTaO4。实验结果表明,钽基的三元材料(Cd2Ta2O4、AgTaO4和 BiTaO4)在可见光下均表现出对甲苯良好的光催化性能。单纯的Bi2WO6和AgVO3由于过快的电子-空穴复合率导致其无法在可见光以及紫外光下降解甲苯气体,但是它们在紫外光照射下,对液体中的甲基蓝的催化活性高于其他材料。Chen等[68]首次通过均相沉淀法在不同pH值条件下制备出CdSnO3·3H2O纳米颗粒。实验表明,在中性以及弱碱性(pH=7, 8)条件下制备的样品,由于其具有更大的比表面积、对光子更好的吸收能力而表现出更优的光催化活性。在254 nm紫外线光催化降解苯的实验中,CdSnO3·3H2O(pH=7)显示出较商品化的二氧化钛P25更强的光催化活性以及更长的使用寿命。
聚合物的石墨相氮化碳(g-C3N4)禁带宽度2.7 eV,是氮化碳的最稳定的同素异形体,被认为是一种理想的光催化剂材料[69]。但是,与Bi2WO6类材料类似,其电子-空穴对复合率过快,为此通常与别的半导体材料进行复合,通过不同材料VB与CB的配合以降低电子-空穴对复合率。目前,对该材料与半导体复合材料的研究主要集中在对液体中有机物的光催化降解[70-72],而对降解气态VOCs的研究较少。Katsumata的团队[65]选择禁带宽度与g-C3N4近乎相同的WO3,通过机械混合法均匀负载到层状的g-C3N4表面(图8(a)和8(b)),并首次探讨了其对乙醛的光催化性能。虽然单纯的WO3催化剂与G2W8(g-C3N4和WO3按照质量比为2:8比例混合的复合材料)在可见光下展现出对乙醛相似的降解率(图8(c)),但是从乙醛光降解矿化后的产物CO2的产量来看(图8(d)),单纯使用WO3的CO2产量(28 %)远低于复合催化剂G2W8的CO2产量(45 %)。这表明单纯WO3虽然可以在可见光下降解乙醛气体,但是仅可以将其氧化为中间产物,其矿化率较低。由于乙醛降解的中间产物大部分依然对人体有害,故可以将大部分乙醛光降解至矿化的G2W8,性能较优,且其中WO3的含量越高,催化效率越高。
Katsumata认为,g-C3N4/WO3复合材料较单纯WO3有较高的光催化性能主要与它的能带结构有关。g-C3N4材料与WO3材料的禁带宽度相似,受光照后两种材料的电子(e-)近乎同时从VB被激发到CB,并在VB上留下空穴(h+)。由于两种材料CB与VB的电位各不相同,g-C3N4的光生电子会转移至WO3的CB(这是因为WO3CB底部的电位+0.5 V( vs. SHE)高于g-C3N4底部的电位-1.13 V(vs. SHE))。类似地,位于WO3材料VB上的空穴会转移到g-C3N4的VB。这种电子-空穴对的转移在可见光催化降解乙醛中被证明是有利的。
3 VOCs催化氧化材料
催化燃烧(氧化)法是一种利用合适的催化剂,使得VOCs气体能在较低的温度下(<300℃)被氧化成CO2与H2O的手段。该方法较直接燃烧法有着能耗低、净化率高以及无二次污染等的优点,被认为是消除VOCs气体的有效方法之一而受到学界的关注[5,13]。催化燃烧的催化剂主要可以分为两类:贵金属负载型催化剂和过渡金属/金属氧化物型催化剂。本文将对近年来用于VOCs催化燃烧技术的催化剂材料的最新研究情况作简要介绍。
3.1 贵金属负载型催化剂
在催化燃烧反应中,贵金属负载型催化剂(铂、钯、金等)是目前最有效的催化剂。针对该类催化剂的研究主要集中在合适催化剂载体的选择以及通过负载手段提高贵金属在载体表面的负载量和分散度,以此提高催化剂活性。
Zhao等[5]通过原位氧化还原反应将钯(Pd)纳米颗粒负载到核壳结构的AlOOH@CoAl-LDHs表面形成AlOOH@Pd-CoAl-LDHs,在600℃空气气氛中经过煅烧后形成Al2O3@Pd-CoAlO微球(Pd-CoAlO-Al)(图9)。Zhao等通过类似的方法还分别制备了CoAlO-S、CoAlO-Al以及Pd-CoAlO-S,以此细致考察了催化剂载体对催化氧化甲苯效果的影响。在催化氧化甲苯的实验中,上述催化剂的反应活性依次为CoAlO-S <CoAlO-Al< Pd-CoAlO-S < Pd-CoAlO-Al(具体的催化剂活性信息见表1)。文中Zhao将其归因于在Al2O3表面均匀分布的Pd-CoAlO纳米片以及其与基底Al2O3之间强烈的作用。此外Zhao特别指出,催化剂表面生成的PdO是促进催化剂反应活性的重要原因。

图8 (a~b)合成g-C3N4/WO3复合材料的TEM图像;(c)光催化前后CH3CHO的浓度与光照时间的关系图;(d)由光催化降解CH3CHO所产生的CO2浓度与光照时间的关系图;(e)g-C3N4/WO3复合材料在可见光催化下降解CH3CHO的机理图[65]

图9 制备Al2O3@Pd-CoAlO催化剂的过程示意图[5]
Wu等[73]分别以HAuCl4、PdCl2为Au源和Pd源,在聚醋酸乙烯酯(PVA)的保护下使用NaBH4作为还原剂,在合成的3D介孔Cr2O3(meso-Cr2O3)表面成功负载上Au纳米颗粒和Pd纳米颗粒。Wu以甲苯作为待降解VOCs物质,对不同Au/Pd含量的催化剂活性以及水分对催化剂活性的影响做了较为详细的研究。当贵金属的负载量达到1.95 %(质量分数)时(Au/Pd的摩尔比为1.0∶2.0),样品1.95Au1Pd2/meso-Cr2O3在空间速度为20 000 mL/(g·h)时测定的T10%、T50%、T90%分别为87 ℃、145 ℃和65 ℃,表现出对甲苯最佳的催化性能(图10(a))。当2.0 %(体积分数)水蒸气被引入反应体系后,甲苯在120 ℃的转化率从19 %左右上升至44 %左右。这主要是因为1.95Au1Pd2/meso-Cr2O3表面的纳米Pd颗粒对水分子有极强的吸附能力,而纳米Au颗粒在水分子存在的情况下能够有效激活O2分子,故当水分子吸附在催化剂表面时,在Pd以及Au的作用下发生如下所示的H-转移反应,产生了氢过氧自由基,因此提高了催化剂的效率。
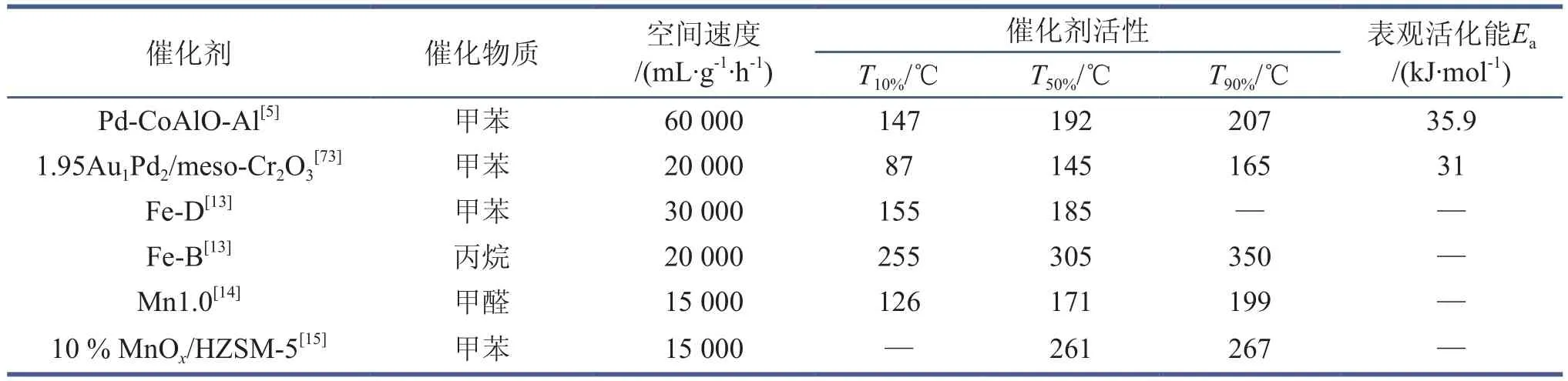
表1 催化剂反应活性以及表观活化能统计表

但是当水蒸气的含量(体积分数)进一步上升至3.0 %、4.0 %时,转化率则分别下降至37 %、34 %。这是因为过多吸附在催化剂表面的水降低了催化剂的活性点位。

图10 (a)在空间速度为20 000 mL/(g·h)时,甲苯转化率与反应温度的关系图 (■meso-Cr2O3;●0.50Au1Pd2/meso-Cr2O3;▲0.97Au1Pd2/meso-Cr2O3;1.95Au1Pd2/meso-Cr2O3;Δ 0.90Au/meso-Cr2O3;◊ 1.00Pd/meso-Cr2O3 );(b)样品1.95Au1Pd2/meso-Cr2O3在空间速度为20 000 mL/(g·h)时,不同水蒸气含量(体积分数分别为2.0 %、3.0 %或4.0 %)对催化氧化甲苯的影响[73]
3.2 过渡金属/金属氧化物型催化剂
虽然上述贵金属催化剂在催化燃烧VOCs上有着优良的活性,但是贵金属自身高昂的价格以及容易失活的特性依旧是其大规模应用的障碍。过渡金属氧化物由于具有多变的价态,快速转移电子的能力,对硫、氯等物质较强的抗性,低廉的成本等优点,被认为是一种替代贵金属催化剂的理想材料。近年来随着纳米技术的快速发展,对过渡金属氧化物的结构、形貌、成分等进行治理以提高其催化氧化活性是研究的热点。
Fe、Cu、Cr和Mn的氧化物是催化燃烧VOCs中一类常见的催化剂材料,对它们性能的研究报道也较多。He等[16]考察了5种金属(Mn、Cu、Fe、Cr、Sn)分别负载在 多孔硅KIT-6上时对氯苯的催化活性。实验结果表明,颗粒的分散性、催化剂的比表面积以及还原能力是影响催化剂活性的主要原因。在He的实验中,催化剂对氯苯的催化活性依次是:Mn/KIT-6 > Cr/KIT-6 >Cu/KIT-6 > Fe/KIT-6 > Sn/KIT-6(以T50%为参考标准),这与催化剂的氧化还原能力强弱是一致的。
Solsona的团队[13]采用不同方法制备了不同结构形貌的α-Fe2O3,并以甲苯和丙烷为对象细致地研究了它们的催化活性。特别值得注意的是,在所有α-Fe2O3催化剂中,两种不同机理形成的介孔催化剂Fe-B和Fe-D分别对丙烷、甲苯展现出了最佳的催化活性。其中,Fe-B的介孔是织构型介孔,由氧化铁纳米晶聚集形成,而Fe-D的介孔则是以介孔二氧化硅KIT-6为模板通过纳米铸造的方式形成(图11)。Solsona在文中认为,造成这种催化性能差异的主要原因是丙烷和甲苯在催化剂表面的催化机理不同。
对于低碳烷烃如丙烷等,在金属氧化物表面发生深度氧化的具体机理目前虽尚不明确,但一般认为与催化剂活性位的还原与再氧化能力有着密切的关系,用Mars-Van Krevelen (MVK) 机理进行阐释。MVK机理指的是反应物(VOCs)与催化剂晶格氧之间的反应机理,主要由两步反应组成:①反应物与催化剂产生氧空位被还原(催化剂被还原);②催化剂被解离吸附的氧补充氧缺位而重新氧化(催化剂被氧化)。Solsona通过建立丙烷氧化与还原能力的关系认为,在催化剂表面发生的还原反应是丙烷完全氧化的限制步骤,因此,已被还原的Fe-B较Fe-D在催化氧化丙烷上有较大优势。对于甲苯的氧化而言,发生的是Rideal-Eley机理,即甲苯分子首先吸附在催化剂表面形成中间体,随后该中间体继续与甲苯分子进行反应。因此,催化剂的表面积以及氧缺陷的相对含量与催化剂的活性直接相关,而催化剂本身的还原能力对催化活性影响不大。Fe-D由于晶桥的存在使催化剂表面相对稳定,还原能力较弱,对于氧化丙烷不占优势,但是XPS分析显示其表面存在有大量的氧缺陷位,因而对甲苯的催化作用明显。
Liu等[14]使用湿法成功制备了Mn掺杂尖晶石型铁氧体,并研究了其对甲醛的催化性能。XPS分析表明,Mn主要以Mn3+和Mn4+存在于尖晶石型铁氧体表面。虽然Mn的引入在一定程度上降低了催化剂的比表面积以及表面吸附的氧含量,造成HCHO吸附量的减少,但是TPR实验表明,Mn的存在有利于尖晶石铁氧体的还原过程,同时显著提高了催化剂表面金属阳离子的氧化能力。在Liu的实验范围内,越高的Mn含量展现出越优异的催化性能(图12)。

图11 低倍TEM图像:(a、e)Fe-A;(b、f)Fe-B;(c、g)Fe-C;(d、h)Fe-D。图(a)的内嵌图为FeOx
Huang的团队[15]通过浸渍的方法将不同浓度的MnOx负载到HZSM-5沸石上,考察了其对甲苯的催化氧化性能。实验表明,MnOx含量为10 %时,样品10%MnOx/HZSM-5展现出了最好的催化活性。TG-MS进一步证明了分散在HZSM-5表面的MnOx可以有效降低催化剂表面焦炭的形成。通常认为载体对于反应是惰性的,但Huang的团队通过对比不同载体材料(SiO2和Al2O3)对催化剂性能的影响,认为HZSM-5对VOCs优良的吸附及捕获能力也是MnOx/HZSM-5具有极强活性的原因之一,此外在HZSM-5表面的Brønsted酸点位对甲苯的催化氧化有促进作用。
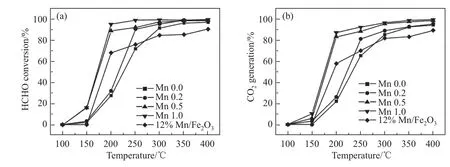
图12 以Mn掺杂尖晶石型铁氧体作为催化剂时,HCHO的转化率(a)和CO2的产生率(b)
4 结语
由于VOCs排放所造成的各类环境问题以及健康问题越来越引起人们的关注,对VOCs的处理已经迫在眉睫。VOCs治理的关键材料是VOCs处理的基础,同时也是决定VOCs治理成败的关键与核心。只有制备出性能更为优越的治理材料,才能更有效地治理VOCs 污染。本文介绍了近年来用于VOCs吸附、光催化降解以及催化燃烧工艺的关键材料的研究情况,认为今后的研究应在目前实验室的水平上,进一步结合实际应用中的工艺水平与条件,开发出可以大规模应用于实际生产的VOCs治理材料。
(2018年10月26日收稿)
猜你喜欢
中华养生保健(2020年9期)2021-01-18 03:12:36
无机化学学报(2019年2期)2019-02-27 06:53:38
石油石化绿色低碳(2019年6期)2019-02-13 09:39:01
陶瓷学报(2019年5期)2019-01-12 09:17:34
三峡大学学报(自然科学版)(2017年1期)2017-03-20 15:30:23
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中国资源综合利用(2016年9期)2016-01-22 08:35:22
中国资源综合利用(2016年4期)2016-01-22 08:27:23
应用化工(2014年7期)2014-08-09 09:20:27
