
沥青质分子聚集体中氢键作用力的研究
2019-03-22 06:01:06代振宇
石油学报(石油加工) 2019年2期
任 强, 龙 军, 代振宇, 周 涵
(中国石化 石油化工科学研究院, 北京 100083)
在石油的生产、储运和加工过程中,沥青质所引起的问题越来越受到人们的重视,目前已成为石油工业中需重点研究和解决的技术问题之一。沥青质的缔合、聚集、沉积以及生焦等问题的最终结果是导致了生产成本的大幅度增加。而这一切的根源,均与沥青质分子形成的超分子结构的聚集体有关。因此,进一步认识沥青质分子的聚集体结构,研究形成沥青质超分子结构的各种作用力,打破沥青质的超分子结构,使沥青质以单分子的形式参与反应,对于重油的加工具有重要的意义。
胶质、沥青质中存在较大的芳香环结构和较多的杂原子,可通过芳香环系的π-π堆积以及与杂原子形成的氢键进行自组装,形成超分子聚集体[1-2]。但是,导致沥青质分子聚集体形成的主要作用力长期以来却颇有争议[3-4]。许多学者认为,氢键是沥青质聚集体形成的主要作用力[5-6],但也有人认为沥青质分子聚集体的形成主要是由分子之间的 π-π 相互作用[7]、极性诱导作用以及静电引力作用等引起的[8-9]。
综上所述,对于沥青质分子形成这种聚集结构的分子间作用力的复杂性,至今没有清晰一致的认识,对沥青质分子聚集体形成的作用力还存在很大的争议, 故对于沥青质分子聚集体的解聚一直是石油加工的难点。笔者采用分子模拟的方法,围绕沥青质分子聚集体中的氢键相互作用进行研究,找到沥青质超分子结构的分子间主要氢键作用力类型及大小,研究沥青质分子聚集体中氢键的本质,掌握氢键对形成沥青质分子聚集体所起的作用,为沥青质聚集体的解聚提供一定的帮助。
1 模型化合物的确定和模拟参数的设置
1.1 模型化合物的确定
由于沥青质的复杂性及分析手段的局限性,长期以来石油行业对沥青质分子的确切结构仍没有形成定论。随着各种先进表征技术的应用,结合微型反应或工业试验数据,对沥青质的认识在逐步加深,尤其近二十年来的研究进展,使得关于沥青质认识得到进一步发展,对沥青质最新的认识将为沥青质分子模型的构建提供有利的参考。在得到元素组成和相对分子质量的基础上,核磁共振法(NMR)和X射线光电子能谱(XPS)分别是目前获得碳氢结构信息和杂原子官能团信息的最有效表征方法。根据实验分析数据和文献[10]方法,确定本研究的沥青质分子和胶质分子的模型化合物如图1所示。图1中灰色的球为C原子,白色的球为H原子,红色的球为O原子,黄色的球为S原子,兰色的球为N原子(下同)。
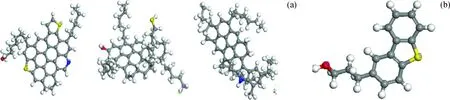
图1 模型化合物分子Fig.1 Model compound molecules(a) Asphaltene molecules; (b) Resin molecules C atom; H atom; S atom; O atom; N atom
1.2 模拟参数的设置
采用Materials Studio 8.0对模型进行分子动力学结构优化时,根据文献[11-12],对于沥青质体系,采用NVT系综,Andersen方法,温度控制在600 K,以确保大分子构象的松弛和越过较高的能垒,力场采用Compass力场,时间步长为1 fs,总模拟时间为500 ps,结果记录文件为每计算500步输出一个结构,截取半径按照小于所建模拟盒子边长的一半取1.35 nm。
在ADF模拟中,分子轨道使用Slater基组TZ2P展开(不含任何Gaussian函数)。对于C、N、O、S原子,冻结了1s轨道[13]。而在自洽场(SCF)计算过程中,计算库仑、交换相关势时,软件自动采用一套包含s、p、d、f、g的Slater函数来拟合分子的电子密度[14]。泛函采用的是BLYP-D3(BJ),BLYP为GGA泛函的一种,D3(BJ)是一种对泛函的长程行为进行“经验参数式”修正的所谓“色散修正”方法[15]。色散修正主要用于弱相互作用体系,例如氢键、超分子。需要进行色散修正的原因主要是因为一般的密度泛函势函数随着离原子核距离的增大衰减过快,因此对距离较远的原子的相互作用描述得不好。经D3(BJ)修正之后,原子间长程相互作用势的衰减形式为C6/(R6+c)[16]。其中c、C6为拟合得到的常数,R为离原子核的距离。简单的说就是呈6次方衰减。结构优化使用的是软件默认的解析梯度方法。
2 结果与讨论
2.1 沥青质分子中氢键的类型及键能大小
2.1.1 沥青质分子中氢键的类型
氢键是分子间作用力的一种,是一种永久偶极之间的作用力。氢键发生在已经以共价键与其它原子键合的H原子与另一个原子之间(X—H…Y),通常发生氢键作用的H原子两边的原子(X、Y)都是电负性较强的原子。
沥青质分子中含有N、S、O等杂原子,具备形成氢键的条件,可以自身形成分子内氢键,也可以与其他的沥青质分子形成分子间氢键。根据其所含杂原子的类型,对不同的沥青质分子进行分子动力学模拟,可分别形成如图2所示的O—H…N氢键、S—H…N氢键、O—H…S氢键、O—H…O氢键、N—H…N氢键、S—H…S氢键等6种类型的氢键。沥青质分子可以与水分子形成氢键,可以与胶质分子形成氢键,也可以与其他类型的沥青质分子形成氢键。石油中只要含有N、S、O等杂原子的化合物,理论上均可与沥青质分子形成氢键,但不管与什么分子形成氢键,其类型均在上述的6种氢键类型之中。
采用Amorphous模块将沥青质分子分别与胶质分子、水分子和沥青质分子放入不同的模拟盒子中,进行分子动力学模拟,考察其形成氢键的类型。结果发现,沥青质分子与沥青质分子、胶质分子及水分子均能够形成O—H…S氢键、O—H…N氢键和O—H…O氢键;而 S—H…N氢键、S—H…S氢键和N—H…N氢键是由沥青质分子分别与沥青质分子、胶质分子形成的;与水分子则无法形成S—H…N氢键、S—H…S氢键和N—H…N氢键。沥青质聚集体中实际存在的氢键类型如表1所示。
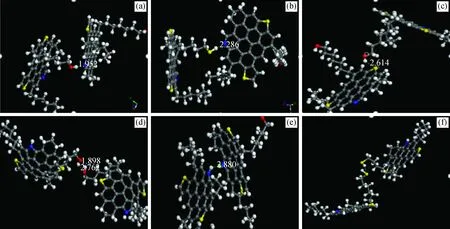
图2 氢键的类型Fig.2 Typical hydrogen bonds(a) O—H…N hydrogen bond; (b) S—H…N hydrogen bond; (c) O—H…S hydrogen bond;(d) O—H…O hydrogen bond; (e) N—H…N hydrogen bond; (f) S—H…S hydrogen bond
2.1.2 沥青质分子中氢键键能的大小
采用量子力学方法对沥青质分子间的相互作用进行研究,可以得到更多的微观结构信息,相关的研究已有报道[17]。为了计算沥青质聚集体中氢键键能的大小,将分子动力学模拟中已经形成氢键的各类型分子结构不变的提取出来,在此基础上再分别进行量子力学计算,得到沥青质分子与沥青质分子、水分子和胶质分子间的各种氢键键能及其他结构参数,如表1所示。氢键键能的计算方法参照文献[18]的模拟方法。
由表1可以看出,沥青质聚集体中氢键的键能在9~39 kJ/mol之间,其中O—H…N氢键键能相对于S—H…N氢键和O—H…S氢键略大。另外,同种类型的氢键,键长越小,其键能相对越大。
图3为沥青质分子与水分子形成的O—H…S氢键,其中图3(a)为形成氢键后分子键长的变化,图3(b)为形成氢键后电子云的分布情况。从图3(a)可以看出,形成氢键后,水分子中的H—O键的键长为0.0981 nm,而水分子单独存在时水分子中的H—O键的键长为0.0961 nm;同样,形成氢键后,沥青质分子中的S—C键的键长为0.1784 nm,而同一沥青质分子中没有形成氢键的S—C键的键长为0.1758 nm,可见,氢键形成后,原分子中的H—O键和S—C键的键长均变大。从图3(b)可以看出,形成氢键后,水分子中形成氢键的H原子的电子云与沥青质分子中形成氢键的S原子的电子云有部分重合,这说明H原子与S原子的价层轨道电子进行了叠加。
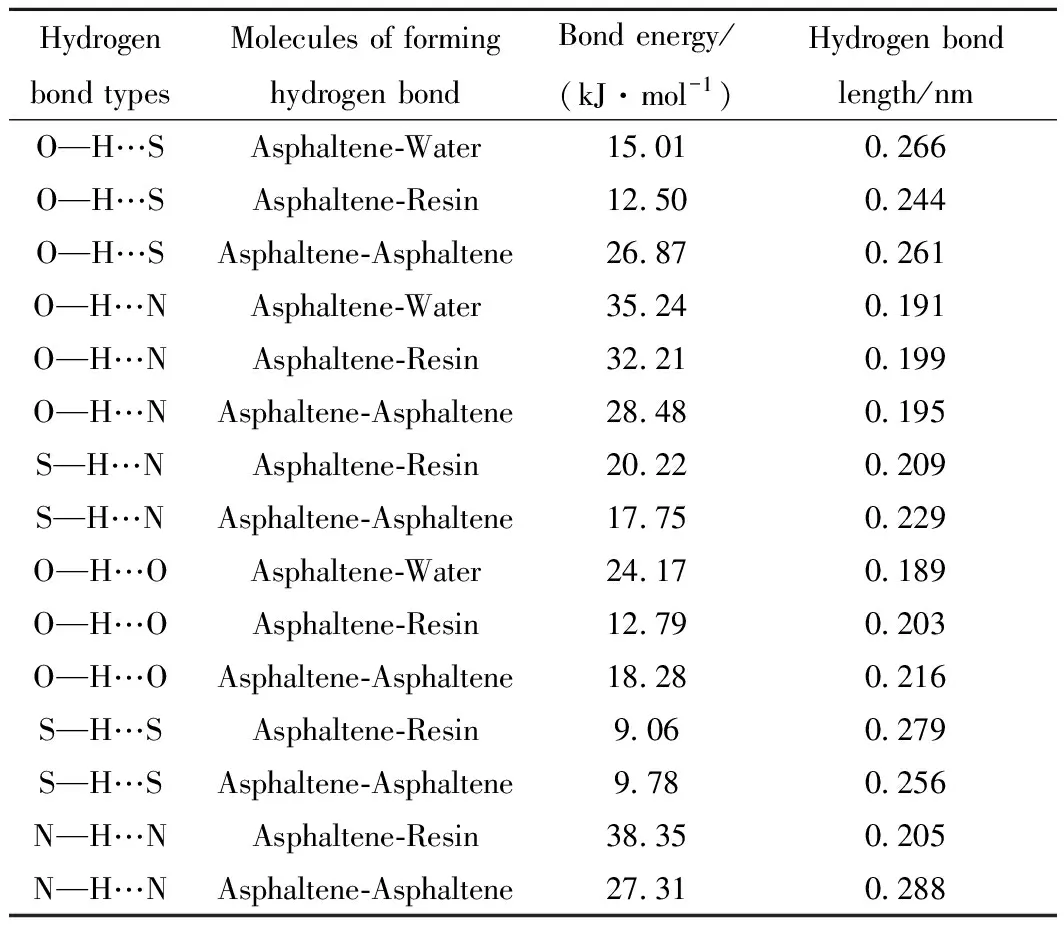
表1 沥青质聚集体中氢键的类型及键能大小Table 1 Typical hydrogen bonds and bond energyin asphaltene aggregates
图4为沥青质分子与水分子形成O—H…S氢键后相关原子的电荷变化情况。可以看出,当水分子和沥青质分子单独存在时,水分子中O原子上的电荷为-0.810e(如图4(a)所示),沥青质分子中S原子上的电荷为-0.168e(如图4(b)所示);当水分子与沥青质分子形成O—H…S氢键后,水分子中O原子上的电荷变为-0.837e,沥青质分子中与水分子形成氢键的S原子上的电荷变为-0.185e(如图4(c)所示)。由此可见,形成氢键后,分子间发生了电荷转移,由于O原子的电负性比S原子更强,故吸引了更多的负电荷,从而使其变得更负,而S原子由于与多个苯环形成了共轭体系,其电荷发生部分转移后,其共轭体系中的原子电荷进行了再分配,从而使其电荷也变的更负,进一步加强了与H原子的结合能力。
由表1可以看出,氢键键能相对较小,其形成和破坏需要的活化能也小,加上形成氢键的空间条件比较灵活,在重油内部分子间和分子内不断运动变化的条件下,氢键仍能不断地断裂和形成,在重油体系中保持一定数量的氢键结合。

图3 沥青质分子与水分子形成的O—H…S氢键Fig.3 O—H…S hydrogen bonds formed between asphaltene molecules and water molecules(a) Change in bond length after hydrogen bonding (nm); (b) Distribution of electron clouds after hydrogen bonding

图4 沥青质分子与水分子形成O—H…S氢键后相关原子的电荷变化Fig.4 Atomic charge changes after formation of O—H…S hydrogen bonds between asphaltene molecules and water molecules(a) Water; (b) Asphaltene; (c) Hydrogen bonds forming between water and asphaltene
由于沥青质分子中会同时含有S、N、O等杂原子,因此,多个沥青质分子间有可能同时形成2个甚至多个氢键。同时,沥青质分子聚集体中会有多个沥青质分子通过π-π相互作用聚集在一起,定义2个沥青质分子间的π-π相互作用方式为“π-π”,3个沥青质分子间的π-π相互作用为“2 π-π”;同时,定义2个沥青质分子间形成的1个O—H…S氢键为“SO氢键”,2个沥青质分子间形成的2个O—H…S氢键为“2SO氢键”,2个沥青质分子间形成的3个O—H…S氢键为“3SO氢键”。
对沥青质分子聚集体中形成的多氢键和多π-π相互作用进行计算,结果显示,分别由1个、2个和3个SO氢键和2π-π形成的相互作用能分别为36.6、69.13和85.00 kJ/mol。由此可以看出,在相同的π-π相互作用能下,随着沥青质聚集体中氢键数量的增加,其分子间的相互作用能增加。因此,沥青质分子中虽然单个氢键键能较小,但聚集体含有多个氢键时,其分子间的作用力会大大增加。故沥青质分子聚集体在多个氢键的共同作用下,分子间作用力较大,要打破这种作用力,使沥青质分子以单分子的形式存在,需要的能量也较大。
2.2 沥青质分子中氢键相互作用的本质
由上述氢键的类型可以看出,形成氢键的2个分子电子结构方面有互补的特性,即接收质子的原子是负电性的,而质子本身带上了正电。图5为2个沥青质分子形成的O—H…N氢键,其中点亮的分子为其中1个沥青质分子,未点亮的分子为另外1个沥青质分子。下面考察电荷转移相互作用的可能性。
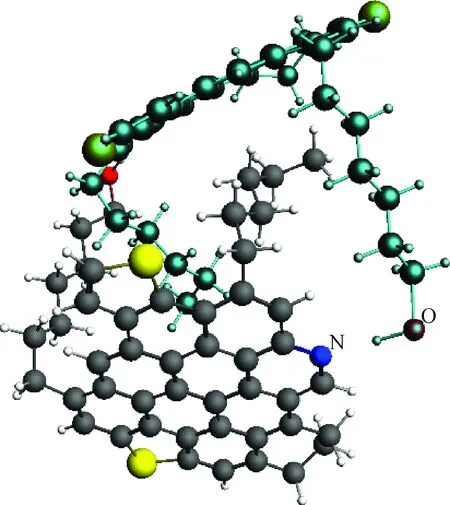
图5 2个沥青质分子形成的O—H…N氢键Fig.5 O—H…N hydrogen bonds formationbetween two asphaltene molecules C atom; H atom; S atom; O atom; N atom
图6为图5所示的沥青质分子二聚体与沥青质分子单体之间的部分轨道关系图。中间为沥青质分子Asphaltene 1和Asphaltene 2形成的二聚体的轨道,两边分别为单体沥青质分子1、沥青质分子2的轨道。图6并未显示完整的单体与二聚体轨道关系,只列举了少数几个轨道的关系,如HOMO(最高占有轨道)、HOMO-1、LUMO(最低空轨道)、LUMO+1等,并且只显示了其中施主、受主效应相关的轨道关系。
左边Asphaltene 1上方的127A就是Asphaltene 1的σ的LUMO,126A 即Asphaltene 1的σ的HOMO;右边Asphaltene 2的前线轨道与Asphaltene 1的类似。Asphaltene 1+Asphaltene 2 上方的251A、252A即沥青质分子二聚体的σ的 HOMO-1和HOMO,上方的2条 253A和254A对应沥青质分子二聚体的σ的2个空轨道LUMO+1和LUMO。

图6 沥青质分子二聚体与沥青质分子单体之间的部分轨道关系Fig.6 Partial orbital relationship between asphaltenemolecule dimer and asphaltene monomer
采用ADF软件对不同的轨道进行分析,可以得到如图6所示二聚体轨道的组分主要来源(见表2)。图6中最高占有轨道 252A 的组分来源于Asphaltene 1的126A和Asphaltene 2的126A,Asphaltene 1的 126A占有绝大多数,而Asphaltene 2的126A则占有很小的比例,说明该二聚体的最高占有轨道主要是由Asphaltene 1的电子组成,Asphaltene 2则贡献了较少的电子。同样,作为HOMO-1的251A的组分来源则与HOMO相反,Asphaltene 2贡献了较多的电子,而Asphaltene 1则贡献了较少的电子。另外,对沥青质分子二聚体的LUMO、LUMO+1轨道253A、254A进行分析,发现LUMO主要由Asphaltene 2和Asphaltene 1共同组成,但Asphaltene 2占据了主要部分,Asphaltene 1只占据了很少的一部分;而LUMO+1与LUMO正好相反,Asphaltene 1占据了主要部分,Asphaltene 2只占据了很少的一部分。由此可见,形成二聚体时2个单体都对二聚体的轨道产生了一定的影响,只是在不同的轨道,某个单体占据主要组分,另一个单体只占有一小部分组分;由此也说明,在形成氢键的过程中,单体间有极少量的电子转移,导致形成弱的次级键。

表2 形成二聚体轨道的组分主要来源Table 2 Main sources of orbital components for dimer
2.3 沥青质分子中氢键能的组成
对形成氢键的沥青质分子二聚体的氢键能ΔE定义如下:
ΔE=Edimer-Emonomer1-Emonomer2
(1)
式(1)中,Edimer为优化得到的沥青质分子二聚体的能量,kJ/mol;Emonomer1和Emonomer2分别为沥青质分子1和沥青质分子2优化得到的能量,kJ/mol。总的键能ΔE在物理含义上包含2个部分:
ΔE=ΔEprep+ΔEint
(2)
式(2)中,ΔEprep为单体从自由状态的结构发生形变,变为二聚体中的结构所需要消耗的能量(该能量一般情况下为正值,因为自由状态的结构能量肯定是局域最小的),kJ/mol;ΔEint为2个形状已经“准备好”的单体,保持结构不变地形成二聚体时释放的能量(该能量一般情况下为负值,否则二聚体能量高于单体能量之和,则无法形成二聚体),kJ/mol。
该氢键体系的相互作用能(ΔEint)在Kohn-Sham分子轨道框架下进行定量的能量分解(Energy decomposition analysis,EDA),分解成静电相互作用能(ΔEelastat)、Pauli 排斥轨道相互作用能(ΔEPauli)、“吸引”轨道相互作用能(ΔEoi)以及色散相互作用能(ΔEdisp)[19]。
ΔEint=ΔEelstat+ΔEPauli+ΔEoi+ΔEdisp
(3)
ΔEelstat为经典概念下单体结构已经完成“形变准备”(即单体已经从自由状态的结构调整成二聚体中的结构),2个单体之间的电子密度还互不影响时的2个沥青质分子单体之间的静电相互作用。电子密度空间分布为2个孤立单体的电子密度直接叠加。通常,这是吸引作用(否则无法形成二聚体)。
Pauli排斥轨道相互作用能ΔEPauli包括占据轨道之间的倾向于破坏稳定的相互作用能,即产生了占据轨道之间的在空间上互相排斥的效果。
轨道相互作用ΔEoi在任何分子轨道理论模型中,包括密度泛函Kohn-Sham理论,都是产生电荷转移的原因所在。电荷转移是由施主的占据轨道和受主的空轨道之间的相互作用,即互相混合所造成。HOMO-LUMO 之间的相互作用,也包括在其中。这部分轨道的相互作用,导致了原子之间出现电荷转移。另外,ΔEoi也导致极化,由于另一个单体的存在,导致某单体自己的空、占轨道之间也在一定程度上互相混合;这部分轨道的混合,导致了单体的极化。
ΔEdisp为色散修正能量。
表3列出了1个只形成O—H…N氢键的沥青质分子二聚体的氢键能ΔE的分解值。
从表3可以看出,键能首先分解为单体的“形变准备”能ΔEprep以及单体之间的相互作用能ΔEint。前者非常小,因为氢键导致的形变量很小,而ΔEint则相对较大。进一步分解ΔEint发现,静电作用能ΔEelstat不提供净的键能,只能部分补偿 Pauli 排斥轨道相互作用能ΔEPauli。如果没有轨道之间的相互作用能ΔEoi,单体将会互相排斥,因此,这2种作用能是相伴而生的。ΔEPauli与ΔEelstat之和的大小反映了分子间的空间相互作用,即位阻效应的强弱。因为ΔEelstat是分子间的吸引力,当吸引到一定的距离时,由于分子中原子间的电子云的排斥作用,2个分子又不能离得太近,故2个分子在一定的空间平衡位置附近保持分子的振荡,因此其反映出了分子在空间上的排布特征。ΔEPauli与ΔEelstat之和可以看作一个相互作用能,即空间相互作用能(ΔEsteric),又叫空间位阻能,其反映的是相邻分子间的空间位阻效应。

表3 沥青质分子二聚体氢键能的分解Table 3 Decomposition of hydrogen bonding energy of asphaltene molecular dimer kJ/mol
从表3还可以看出,在沥青质分子二聚体形成的氢键中,空间相互作用能ΔEsteric=24.08 kJ/mol,相对于ΔEoi和ΔEdisp较小,因此,在氢键中起主要作用的是轨道相互作用和色散作用。
3 结 论
(1)沥青质分子由于含有一定的杂原子,能够与其它含有杂原子的分子形成6种类型的氢键。沥青质分子形成的单个氢键键能的大小为9~39 kJ/mol。
(2)沥青质分子聚集体虽然形成的单个氢键键能较小,但聚集体中含有多个氢键时,其分子间的作用力会大幅增加。
(3)沥青质分子形成氢键的本质是由于H原子与杂原子的价层轨道电子进行了叠加形成的,沥青质分子间有极少量的电子转移,导致形成弱的次级键。
(4)沥青质分子形成的氢键中起主要作用的是轨道相互作用和色散作用。
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
合成化学(2024年3期)2024-03-23 00:56:44
合成化学(2023年12期)2024-01-02 01:02:18
河南工业大学学报(自然科学版)(2021年6期)2022-01-26 06:36:08
中学生数理化·高三版(2016年2期)2016-09-10 07:22:44
中国卫生(2014年5期)2014-11-10 02:11:32
石油化工(2014年1期)2014-06-07 05:57:08
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
无机化学学报(2014年1期)2014-02-28 17:30:06
机械与电子(2014年2期)2014-02-28 02:07:43
