
吲哚类Src/IGF-1R激酶双靶点抑制剂的分子对接和分子动力学研究
2019-03-14 02:35郑晓杰马少杰吴文娟郑康成
广东药科大学学报 2019年1期
郑晓杰,马少杰,吴文娟,郑康成
(1.广东药科大学药学院,广东 广州 510006; 2.中山大学化学与化工学院,广东 广州 510275)
胰岛素样生长因子-1受体(IGF-1R)是一种跨膜受体糖蛋白,在各种类型细胞中均有表达。研究发现,IGF-1R的信号转导途径与肿瘤的发生、发展、增生、转移、血管生成和凋亡等密切相关[1-3],在多种人体实体瘤如肝癌、淋巴癌、头颈鳞癌等中均有异常表达[4]。因此,IGF-1R成为治疗肿瘤的有效靶标而受到人们的广泛关注,各种小分子IGF-1R抑制剂得到开发并已用于试验阶段[5]。
c-Src作为非受体酪氨酸激酶,调节着IGF-1R和下游级联信号通路的信号,在癌细胞的多个信号转导通路中起着重要的作用。Src的过度表达会导致人体恶性肿瘤如结肠癌、乳腺癌、胰腺癌和脑癌等的产生[6-9],因此,特异性地阻断其信号转导可抑制肿瘤的发生和发展。目前,关于Src激酶抑制剂的研究还在不断进行,小分子抑制剂如XL-999、AZD-0530、KX010107和CGP70630已进入临床试验阶段[10-11]。然而,肿瘤的发生发展是错综复杂的,肿瘤细胞中的信号相互交错、相互影响。因此,大多数单靶点抑制剂在长期使用后易引发机体的耐药性[12],而双靶点或多靶点抑制剂可同时作用于某一疾病的多个分子靶点,且能有效克服单靶点药物易引起的耐药性,在肿瘤的治疗方面具有更显著的优势。因此,双靶点或多靶点抑制剂是当前新药研发中的热点领域[13]。研究发现,Src可促进IGF-1R的磷酸化作用且两者具有显著的序列相似性[14-15]。因此,设计新型高效的Src/IGF-1R双靶点抑制剂是治疗癌症的重要方向。Conrad Kunick研究小组合成了一系列新型的吲哚类c-Src/IGF-1R双靶点抑制剂[16],这些化合物的活性测试表明这些化合物可同时对c-Src和IGF-1R激酶产生较强的抑制作用,但是这些化合物与c-Src和IGF-1R之间的作用模式还未有研究,因此,可以通过计算机辅助药物设计来进行理论研究。
对受体与配体间的作用机制进行详细的理论研究将有助于简化药物的研发过程,提高新药研发的速度[17]。分子对接[18-20]可预测配体在受体活性口袋中可能的空间构象,而分子动力学模拟(MD)[21-22]则可以形象地展示分子的构象变化及结构波动情况,从分子水平上描绘受体蛋白与抑制剂的相互作用细节,因此,两者的联合应用有助于更深入地研究配体和受体相互作用的结构特点。本文采用吲哚类衍生物与c-Src和IGF-1R激酶蛋白进行分子对接和分子模拟研究,以探讨抑制剂与两受体间最可能的空间构象及详细的分子作用机制,研究结果将为进一步设计与合成新型高效的双靶点c-Src/IGF-1R抑制剂提供参考。
1 计算和实验方法
1.1 分子对接
采用Sybyl 6.9[23]软件中的Sketch Molecule模块构建小分子和Computer/Minimize模块进行能量优化。在优化过程中赋予分子Tripos立场以及Gasteiger-Hückel电荷,然后,采用Powell能量梯度法,收敛条件为0.42 kJ/(mol·nm),优化1 000步,得到分子的最低能量构象作为对接的配体起始构象。Src和IGF-1R激酶的晶体结构从PDB数据库中获得,PDB代码分别为2BDF(Src)和2ZM3(IGF-1R)。对接之前,去除晶体结构中的水分子和原配体,加氢及赋Kollman电荷[24],并对氢原子位置进行局部能量优化。使用Surflex-Dock模块进行分子对接计算,设置参数Threshold和Bloat分别为0.5和0,以生成一个包含整个疏水口袋区域的Protomol用作对接原型分子,并采用Cscore评价小分子不同构象的对接结果。从每个小分子最终得到的20个最优构象中选取具有最高对接得分及其对接构象与原配体相似的结构作为动力学模拟的初始结构。
1.2 分子动力学模拟
采用AMBER 9.0[25]软件包对对接得到的复合物进行分子动力学模拟。首先,用Gaussian 09软件包在B3LYP/6-31G(d)水平下[26-28]计算小分子的静电势(ESP),然后用Antechamber模块中的RESP分析ESP的结果,得到小分子的原子电荷。MD模拟中,激酶蛋白采用AMBER ff03立场,小分子配体立场采用GAFF[29-30]。溶剂采用TIP3P水模型,模拟体系均添加1.2 nm的水分子层[31-32],并加上抗衡离子Na+使体系呈电中性。MD模拟前须对体系进行能量优化,所有体系先用最陡下降法优化2 000步,再用共轭梯度法优化3 000步以消除分子间的高能碰撞。然后约束溶质对体系进行200 ps的升温过程,使体系温度从0 K逐渐升高到300 K,然后进行12 ns的无约束恒温(300 K)恒压(1.013×105Pa)[33]动力学模拟。在整个模拟过程中使用PME方法计算长程静电相互作用[34],SHAKE算法用来限制所有含氢键的伸缩振动,模拟步长为2 fs[35],非键相互作用截断值为0.8 nm,每500步记录1次坐标轨迹。
1.3 结合自由能的计算
从模拟体系的最后2 ns进行取样,每隔10 ps提取1次构象,共得到200个构象用于结合能的计算。结合自由能(ΔGbind)计算采用MM-PBSA法[36-38],计算公式为:
ΔGbind=ΔGcomplex-(ΔGprotein+ΔGligand)=ΔGMM+ΔGsol-TΔS
(1)
式中:ΔGMM是真空下分子力学自由能,ΔGsol是溶剂化自由能,TΔS是熵效应对体系自由能的贡献值。
ΔGMM由范德华能量(ΔGvdw)和静电能(ΔGele)组成:
ΔGMM=ΔGvdw+ΔGele
(2)
溶剂化自由能(ΔGsol)是静电溶剂化的总和,包含极性溶剂化自由能(ΔGele,sol)和非极性溶剂化自由能(ΔGnonpol,sol):
ΔGsol=ΔGele,sol+ΔGnonpol,sol
(3)
式中:ΔGele,sol通过求解有限差分Poisson-Boltzmann (PB)方程得到,溶质和溶剂的介电常数分别设为1.0和80.0。而ΔGnonpol,sol采用溶剂可及表面(SASA)方法计算:
ΔGnonpol,sol=γSASA+β
(4)
式中:γ=3.012 5 kJ/(mol·nm2);β=0。由于熵效应的计算极其耗时且误差较大,还考虑了在对接研究中不同复合物的熵相同,因而忽略了熵部分的计算[39-41]。
1.4 自由能的分解
为了进一步探究这两个激酶靶点与抑制剂结合模式的异同,使用MM/GBSA方法把结合自由能分解到每个残基上[42],每个残基的结合自由能贡献分解为以下4个能量部分:范德华相互作用能(ΔGvdw),静电相互作用能(ΔGele),极性溶剂化能(ΔGele,sol)和非极性溶剂化能(ΔGnonpol,sol),即:
ΔGinhibitor-residue=ΔGvdw+ΔGele+ΔGele,sol+ΔGnonpol,sol
(5)
通过能量分解可了解残基对小分子与激酶作用的贡献,进而探索它们的作用机理,为新的分子设计提供理论指导。
2 结果与讨论
2.1 对接结果
选取结构上具有相同骨架的13个吲哚类Src/IGF-1R抑制剂为研究对象[16],其结构如图1所示,化合物的活性值列于表1。Surflex-Dock对接采用CScore打分函数,选择分数最高的配体构象作为最后对接结果,用于后续的分子模拟。
13个吲哚类化合物均对接到Src和IGF-1R激酶的活性口袋中(图2),其对接得分和实验活性值列于表1。分析对接分值和实验活性值,得到相关系数分别为R=0.800(Src)和R=0.891(IGF-1R),表明对接得分与抑制活性密切相关,对接得分越高,对Src和IGF-1R激酶的抑制活性越高。另外,从图2可见,13个吲哚类化合物在Src和IGF-1R激酶的活性口袋中的对接取向非常相似,说明该类化合物在受体中采用了相似的结合模式。为了进一步探索化合物与受体间的相互作用机理,选取了对Src和IGF-1R激酶具有最高活性和最低活性化合物7p和7h进行深入的对接分析和进一步的MD研究。
图3和图4为化合物7p和7h分别与c-Src和IGF-1R激酶的对接图,可见两个化合物均对接进入激酶的ATP结合位点,并与激酶活性区域的氨基酸残基发生多种相互作用。
在图3中,化合物7p和7h的吲哚环处于Src活性口袋的入口处并靠近激酶的铰链区,环上N1-H均能与Ala390上的羧基氧形成氢键(0.27 nm),同时,O13和O15还可与残基Lys295和Thr338形成长度分别为0.28 nm 和0.27 nm的氢键。此外,7p的O14及R2基团上O17和O20还可与Asp404,Ser345和Thr354形成额外的3个氢键,长度分别为0.34、0.30、0.30 nm,而7h的氧乙酰肼由于部分发生移动使O14远离Asp404及R2基团为氢原子而未能形成更多氢键。明显可见,7p与Src形成增强的氢键作用是其活性比7h更强的重要原因。表1可见,R2基团上(特别是17和20位)连接有电负性大的原子的化合物如7m-7p,其Src的抑制活性都是较高的。可推断,较长的且连接有氢键受体原子(如O或N等)的R2基团可提高对Src的抑制活性。

表1 吲哚类化合物的结构、活性和对接得分Table 1 Structures,activities and docking scores of indole compounds

a.吲哚类衍生物; b.化合物7p; c.化合物7h。
图1吲哚类化合物的结构式
Figure1Structures of indole compounds
除了氢键作用外,化合物还通过与Src激酶的疏水作用来稳定它们在激酶中的结合构象。图3a可见,7p的联苯部分刚好位于Src激酶的N-端区域(Leu273,Gly274)和C-端区域(Leu393,Ser345)的夹心区并与这些残基发生强烈的疏水相互作用。化合物7h的末端苯环C被疏水性氨基酸Val281、Ala293、Met341及Leu393包围(图3b),但7h的疏水作用比7p的较弱。从表1也可看到,R2基团上有苯环D可与C环形成联苯结构的化合物,都具有与7p相似的疏水作用,其大多为活性较高的化合物,如7m-7p。通过比较化合物7p和7h与Src的相互作用,可知氢键作用和疏水作用的差异是导致化合物活性区别的主要原因。
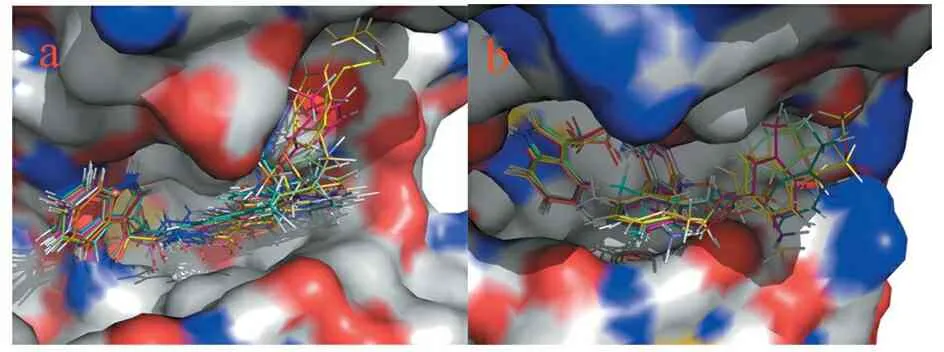
图2吲哚类化合物与Src(a)和IGF-1R(b)激酶的对接构象
Figure2The binding conformations of 13 indole compounds at the active sites of Src(a) and IGF-1R(b)
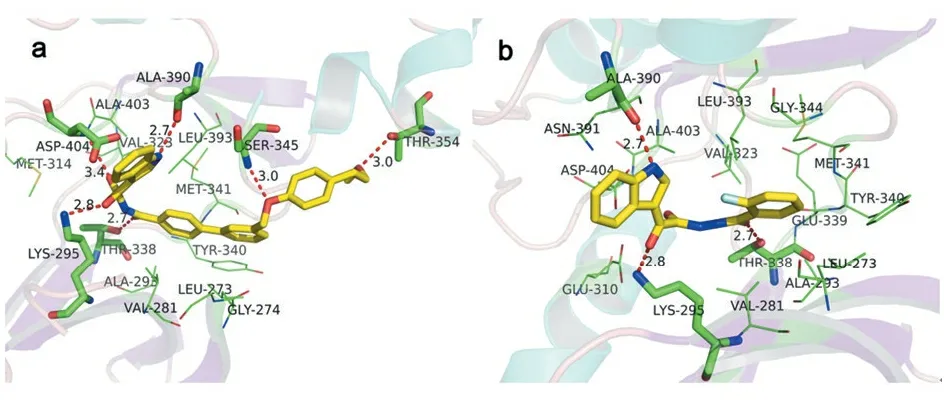
图3化合物7p(a)和7h(b)在Src激酶活性位点的作用模式(氢键用红色虚线表示)
Figure3Interactions between the Src active binding site and compounds 7p(a) and 7h(b) (Hydrogen bonds are depicted as red dotted lines)
图4可见,两个化合物在IGF-1R激酶中的结合构象与c-Src中极其相似,暗示化合物与两种激酶具有相似的作用机制。化合物的吲哚环同样处于IGF-1R激酶活性口袋的入口处,环上的N1H、O15和N11分别与Asp1153、Met1082和Glu1080形成3个氢键,同时,7p中的O13还可与Lys1033形成1个额外氢键,而化合物7h的对接构象相对于7p发生了偏移,导致O13无法与Lys1033形成氢键。此外,7p的联苯部分也处于IGF-1R激酶的N-端(Leu1081,Gly1085)和C-端(Leu1005,Gly1006)的夹心区域,与残基存在大量的疏水作用,其末端醚氧基团伸出活性口袋暴露在溶剂区域,而7h(R2基团为氢原子),由于没有联苯结构导致疏水作用大大减弱。所以,在IGF-1R激酶中,化合物7p比7h增强的氢键作用和疏水作用也是造成其活性不同的重要原因。相比Src,7p与IGF-1R激酶的氢键作用比7h增加没那么多,故其疏水作用的增强更占优势。表1中R2基团有苯环D的化合物,也大多为IGF-1R活性较高的,如7m-7p。
综上所述,对接结果表明吲哚类化合物对Src和IGF-1R激酶高的抑制活性是由于ATP结合位点处形成更多的氢键作用和更强的疏水作用的协同作用导致的。

图4化合物7p(a)和7h(b)在IGF-1R激酶活性位点的作用模式(氢键用红色虚线表示)
Figure4Interactions between the IGF-1R active binding site and compounds 7p(a) and 7h(b) (Hydrogen bonds are depicted as red dotted lines)
2.2 Src与IGF-1R激酶蛋白的结构比较
为了比较两激酶蛋白的相似性,使用http://cl.sdsc.edu/对两蛋白Src(2BDF)和IGF-1R(2ZM3)晶体结构进行叠合,叠合结果列于表2和图5。表2可见,两激酶蛋白叠合的RMSD值和标准分数分别为0.18 nm和7.02 nm,蛋白序列36.00%是相同的,表明Src与IGF-1R两蛋白链相似度较高。图5中,化合物7p的结合位点氨基酸用黑色方框显示,而黑色阴影表示两激酶的序列ID是相同的,由此推断Src与IGF-1R激酶蛋白的对接区域可能有几乎相同的活性位点。
表2 Src(2BDF) and IGF-1R (2ZM3)晶体结构的比较
Table2Comparison of the crystal structures of Src (2BDF) and IGF-1R (2ZM3)

比较的蛋白2BDF-2ZM3RMSD值/nm0.18Z-打分值7.02蛋白序列一致性/%36.00对齐/缺失的位置245/30
2BDF与2ZM3的三维叠加图如图6所示,可见化合物7p与Src的结合构象和相互作用与IGF-1R激酶蛋白极其相似,结合位点处相互作用的关键氨基酸在两个激酶受体中都能很好地叠合,如Gly274/Gly1006、Val281/Val1013、Ala293/Ala1031、Lys295/Lys1033、Met314/Met1054、Val323/Val1063、Thr338/Met1079、Tyr340/Leu1081、Met341/Met1082、Asp404/Asp1153,表明这些吲哚类化合物既对Src激酶有抑制作用,也对IGF-1R激酶有相似的抑制效果。
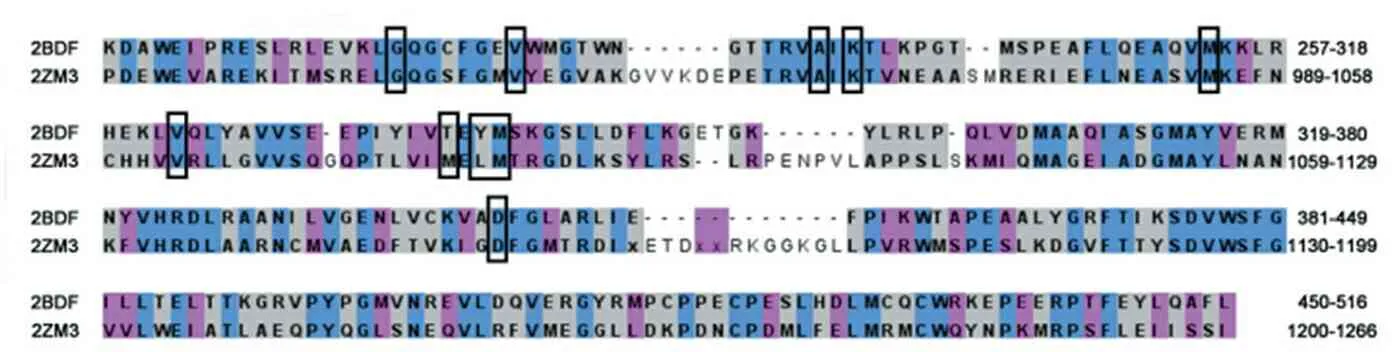
注:浅蓝色区域表示单个列中的氨基酸残基在序列比对中是相同的,虚线表示氨基酸残基的缺失,化合物7p的结合位点用黑色矩形标记。
图52BDF(Src) and 2ZM3(IGF-1R)序列叠合图
Figure5Superposition of the sequences of 2BDF (Src) and 2ZM3 (IGF-1R)
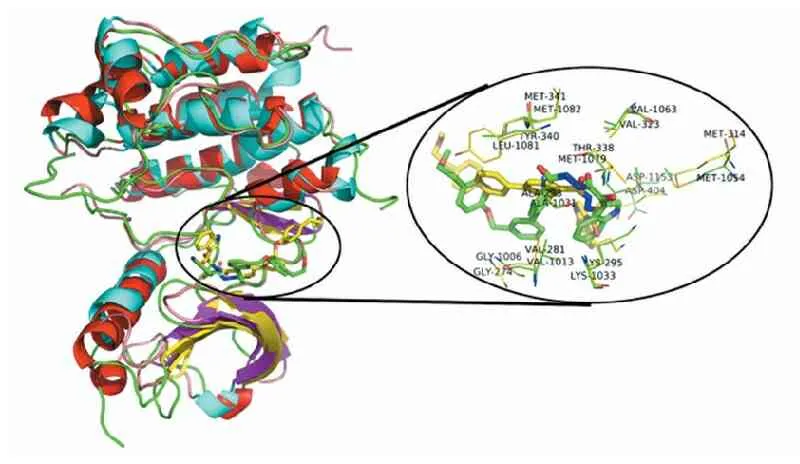
注:投影突显化合物7p(棍状)在活性位点的结构(黄色分子为Src,蓝色为IGF-1R)。
图6Src(2BDF)和IGF-1R(2ZM3)的三维叠合图
Figure6Structure alignment of Src (2BDF) and IGF-1R (2ZM3)
2.3 分子动力学模拟
为了进一步研究配体-受体间的相互作用,对4个对接体系(7p-2BDF,7p-2ZM3,7h-2BDF,7h-2ZM3)进行12 ns的动力学模拟,用蛋白质骨架的均方根偏差(RMSD)衡量各体系的稳定性(图7)。可见,4个体系都在初始2 ns的模拟时间后趋于稳定,RMSD值分别保持在0.22、0.23、0.20、0.22 nm附近,表明模拟过程中各复合物体系达到稳定状态。同时,把各体系对接初始结构和MD最后2 ns的平均结构进行叠加(图8),发现对接结构与MD平均结构位于相同的结合位点,除存在微小的位置偏移外,均能较好地重叠,验证了对接模式的稳定性和合理性。
为描述结合过程中受体氨基酸的贡献情况,计算了各复合物体系蛋白中所有氨基酸残基的均方根波动值(RMSF),结果如图9所示。可见,高活性化合物7p的复合物7p-2BDF和7p-2ZM3中绝大多数残基RMSF值低于低活性的7h复合物,尤其在体系活性口袋区域的氨基酸(Src残基范围:273-295,314-323,338-354,390-393 和403-405;IGF-1R残基范围:1005-1008,1031-1033,1050-1063,1079-1085和1142-1154)的RMSF值比7h明显降低,表明两激酶受体对7p比7h具有更高的亲和力。同时,氢键作用是影响小分子和受体结合的重要因素,故对4个复合物体系的动力学过程进行氢键分析,结果列于表3。可见,7p-2BDF复合物中,抑制剂7p原先在对接构象中与Lys295、Thr338、Ser345、Ala390、Asp404形成的5个氢键重新出现,然而由于其末端醚氧基团产生较大的位置变化,导致O20与Thr354间的氢键消失,但却能与Gln275形成长度相近的氢键相互作用。由图8b可知,在复合物7h-2BDF中,7h的吲哚环及C环在模拟过程中发生了偏移,导致其与Ala390和Thr338的氢键消失,而仅能与Lys295形成1个氢键。然而,复合物7p-2ZM3和7h-2ZM3在MD过程中形成的氢键与对接结果完全相符,表明模拟过程中化合物在IGF-1R激酶口袋中的结合构象比在Src中的稳定。
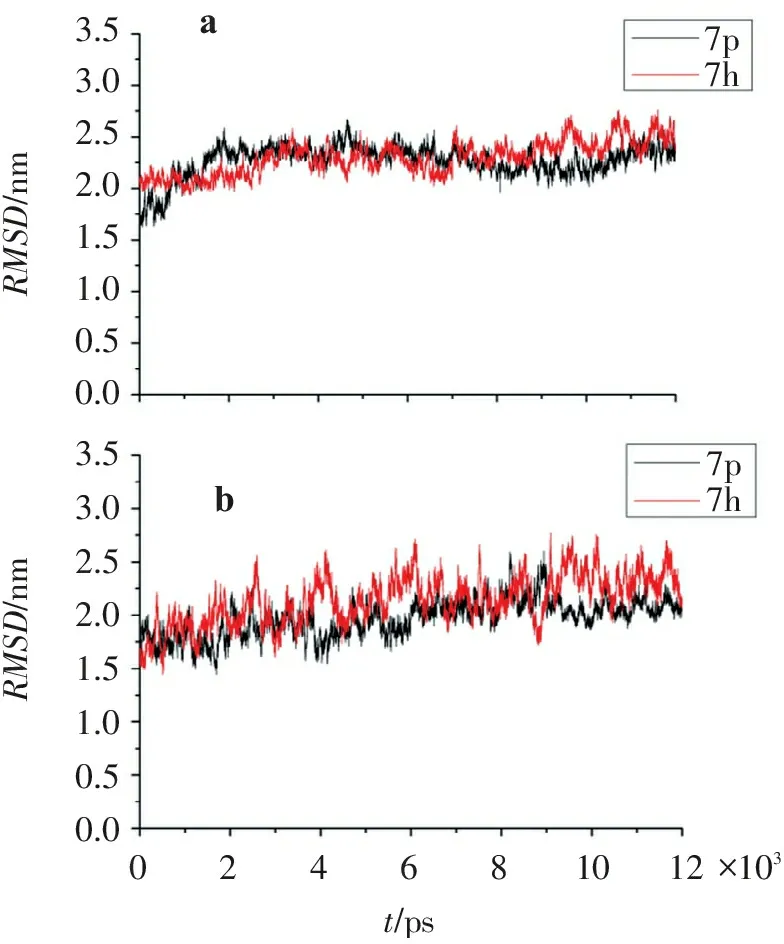
3.53.02.52.01.51.00.50.03.53.02.52.01.51.00.50.0RMSD/nmRMSD/nm7p7h7p7h024681012?103t/psab
a.Src-7p和Src-7h复合物;b.IGF-1R-7p和IGF-1R-7h复合物。
图7Src和IGF-1R复合物体系中RMSD值随模拟时间的变化图
Figure7The rmsd values vs. simulated time in Src and IGF-1R complexes
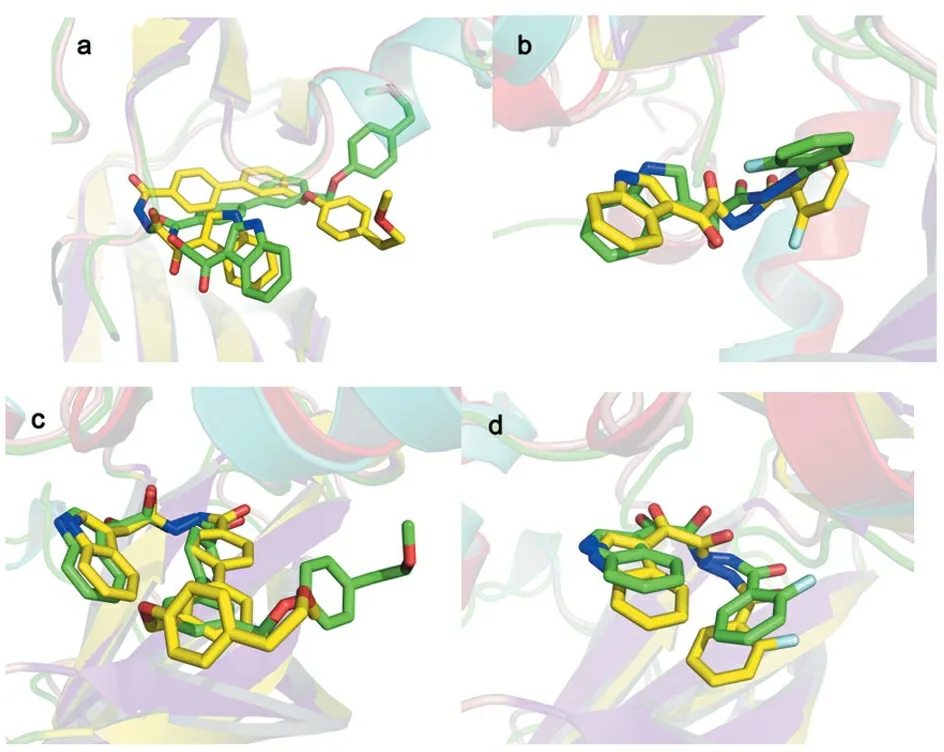
a.Src-7p复合物;b.Src-7h复合物;c.IGF-1R-7p复合物;d.IGF-1R-7h复合物。
图8Src和IGF-1R复合物的初始结构(黄色)和MD模拟平均结构(绿色)的叠加
Figure8Superimposition of the average structure of Src and IGF-1R complex from the last 2 ns of the MD simulation (magenta) and the initial structure (yellow)
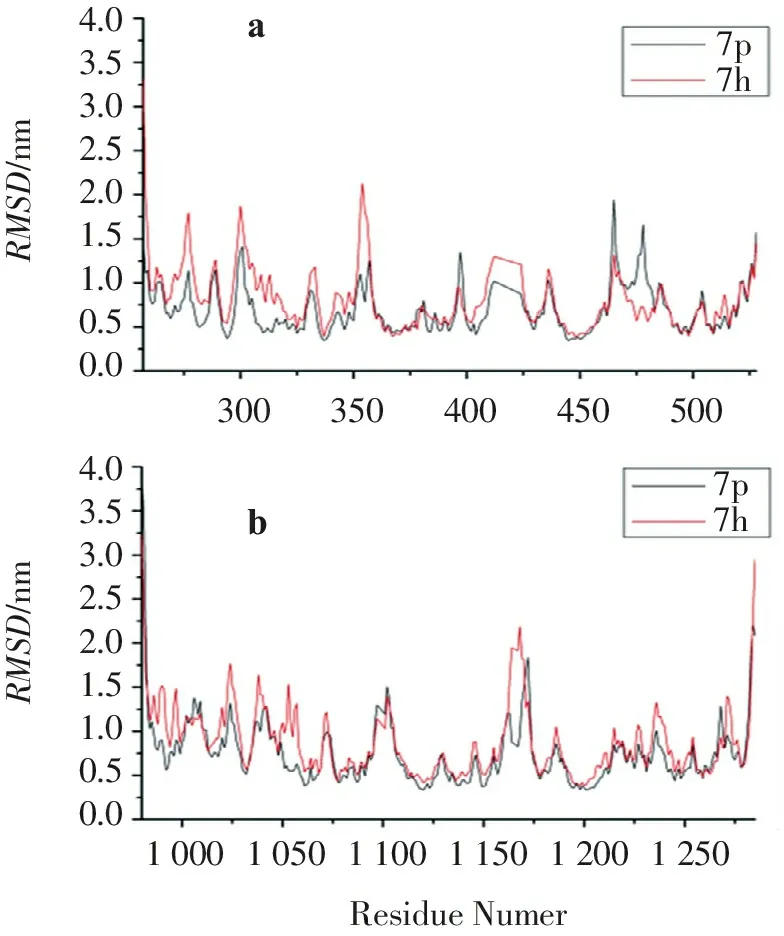
7p7h7p7h4.03.53.02.52.01.51.00.50.04.03.53.02.52.01.51.00.50.0RMSD/nmRMSD/nmab300350400450500ResidueNumer100010501100115012001250
a.Src-7p和Src-7h复合物;b.IGF-1R-7p和IGF-1R-7h复合物。
图9Src和IGF-1R复合物中蛋白质残基的RMSF值
Figure9The RMSFs of each residue of the protein for four Src and IGF-1R complexes
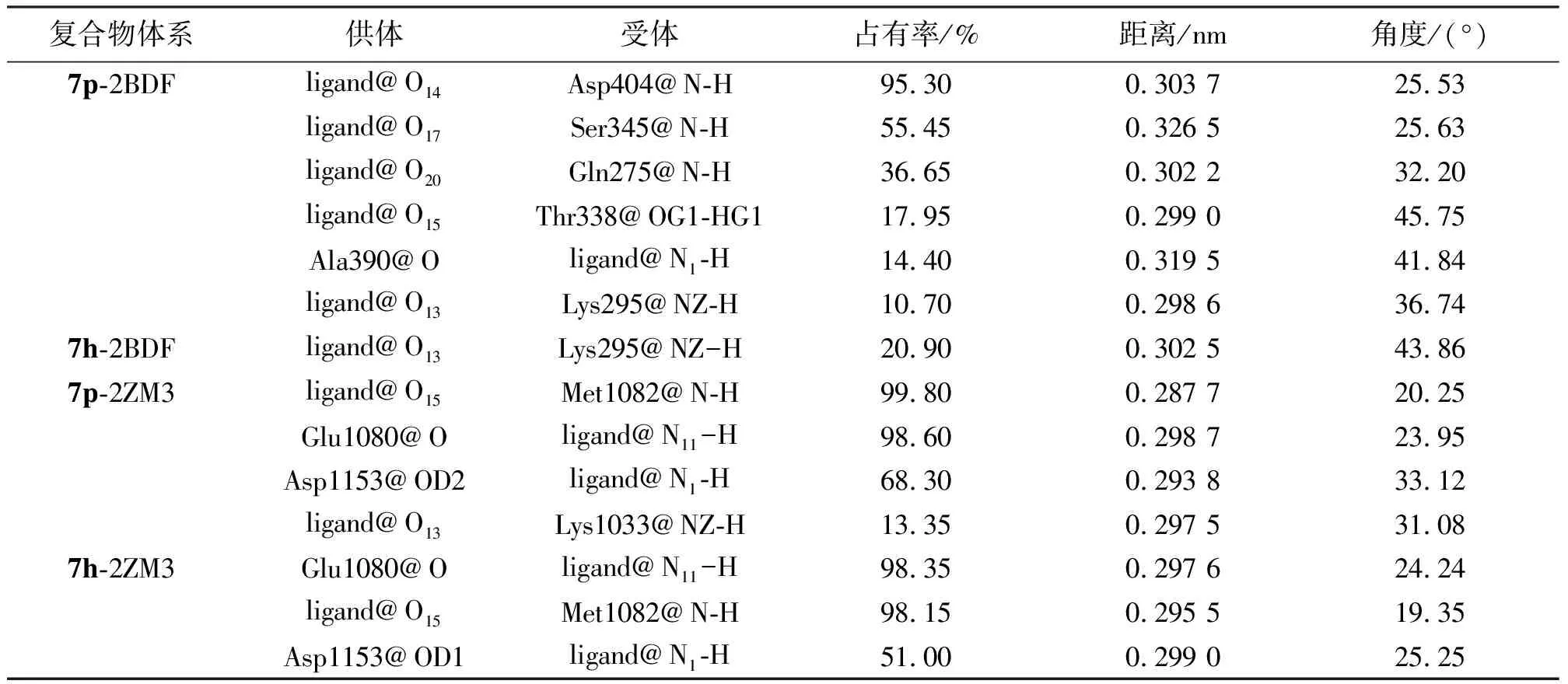
表3 MD过程中的氢键分析Table 3 Hydrogen bonds analysis from MD
2.4 结合自由能分析
为更进一步探究抑制剂与两激酶受体间的相互作用,使用MM-PBSA方法计算4个体系的结合自由能,并将结果列于表4。可见,7p-2BDF和7p-2ZM3体系的结合自由能(-199.45、-148.95 kJ/mol)均比7h-2BDF和7h-2ZM3(-108.45、-106.86 kJ/mol)要高,故7p与两受体的亲和力均比7h要高,可见化合物与受体的结合亲和力和它们的实验活性顺序一致。此外,从表4中还可见,ΔGvdw对系统的结合能有显著贡献,静电相互作用ΔGele的影响也不容忽视,7p-2BDF的ΔGele(-77.86 kJ/mol)比7h-2BDF(-27.99 kJ/mol)高很多,表明7p与Src的氢键作用比7h强得多,而7p-2ZM3的ΔGele(-80.71 kJ/mol)比7h-2ZM3(-76.94 kJ/mol)只稍高一点,说明7p与IGF-1R的氢键作用与7h相关不大,而它们之间的ΔGvdw(疏水作用)都相差较大,这些结果与前面对接的分析相吻合。同时,ΔGnonpol,sol也对结合能有一些贡献,而ΔGele,sol对配体和受体之间相互作用不利。复合物7p-2BDF和7p-2ZM3的ΔGvdw和ΔGele的加和值(-354.17、-271.75 kJ/mol)比7h-2BDF和7h-2ZM3(-192.80、-205.81 kJ/mol)要高得多,表明疏水作用和静电相互作用是维持复合物稳定性的重要因素,这也与前面对接的结论相一致。
为了比较Src和IGF-1R各氨基酸残基对吲哚类衍生物结合自由能贡献的大小,用GBSA方法将结合自由能分解到配体0.6 nm范围内的每个氨基酸上,将值大于8.368 kJ/mol的残基标示于图10中。如图10a所示,在Src的两个复合物体系中,对结合自由能贡献较大的残基为Leu273、Val281、Ala293、Lys295、Val323、Thr338、Met341、Ser345、Leu393、Ala403、Asp404,由于这些残基绝大多数是非极性的,表明这些残基与配体间有强烈的范德华相互作用。残基Leu273、Gly274、Val281、Leu393、Ser345和Asp404对化合物7p的贡献远高于7h,这是由于7p的联苯部分可与这些氨基酸形成强烈的范德华作用,而且Asp404和Ser345能与7p形成稳定的氢键,而与7h不能形成氢键。同时,Lys295在两个体系中都具有较高的能量,这与Lys295可与两个抑制剂都能形成氢键有关。由此可推断疏水相互作用及与Asp404和Ser345的氢键作用是影响Src结合亲和力的重要因素。而在IGF-1R受体中,起关键作用的氨基酸为Leu1005、Val1013、Val1063、Met1079、Glu1080、Leu1081、Met1082、Met1142、Asp1153,同样可推断范德华相互作用在结合自由能中起着主要作用,同时,Glu1080、Met1082和Asp1153与7p和7h间的氢键作用在化合物对IGF-1R的连接中也起着重要的作用。此外,IGF-1R中绝大部分的残基对7p的能量贡献高于7h(图10b),表明IGF-1R对7p的亲和力高于7h,这一结果与两个化合物的实验活性大小相一致。综上所述,疏水相互作用是影响IGF-1R结合亲和力的重要因素。
表4 4个复合物系统的结合自由能
Table4The binding free energy of four systems ΔG/(kJ·mol-1)

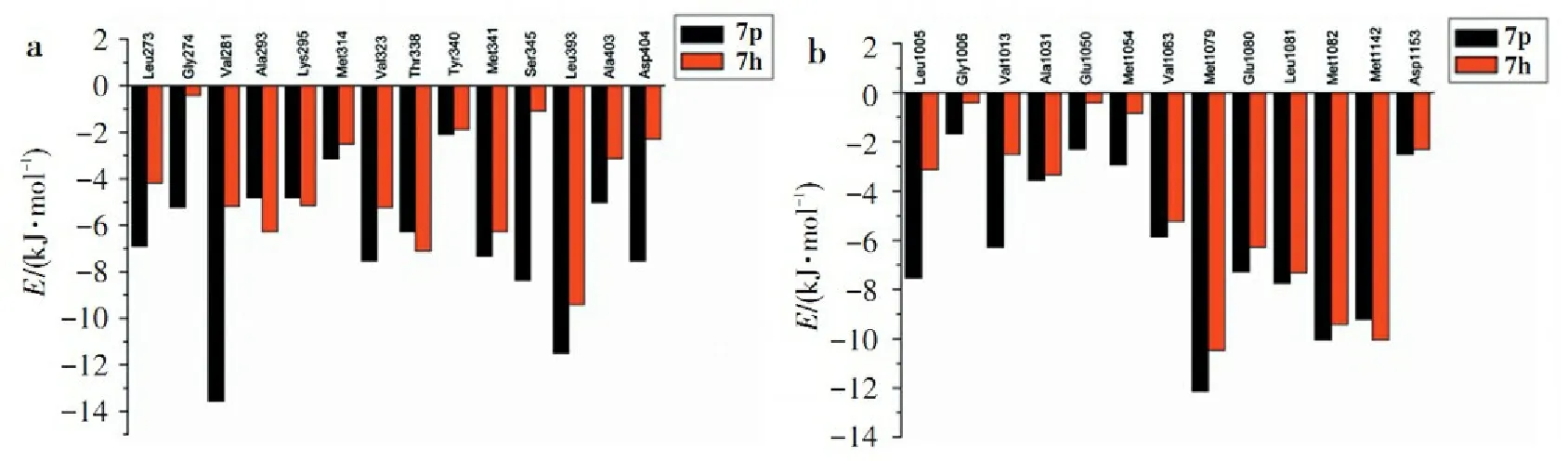
a.Src-7p和Src-7h复合物;b.IGF-1R-7p和IGF-1R-7h复合物。
图104个复合物体系的能量分解图
Figure10Free energy decomposition plots for four systems
3 结论
本文用分子对接和分子动力学模拟的计算方法对新型的吲哚类化合物与Src和IGF-1R激酶受体的相互作用进行了研究。对接结果揭示抑制剂在两个激酶中的结合模式非常相似,对7p的结合力均高于7h,预示两个受体对前者有更高的酶活力,这与实验报道的结果一致。分子动力学模拟和结合自由能计算的结果确证了抑制剂和受体间结合模式的合理性,阐明了疏水作用和氢键作用在受体-配体结合稳定性中所发挥的重要作用,并得到吲哚类化合物与Src和IGF-1R激酶相互作用的关键氨基酸。本文结果可为新型高效的Src/IGF-1R双靶点抑制剂的设计提供重要的理论指导。
猜你喜欢
高中数理化(2022年14期)2022-08-15
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
昆明医科大学学报(2020年12期)2021-01-26
山东化工(2019年11期)2019-06-26
原子与分子物理学报(2019年5期)2019-04-28
中学化学(2015年12期)2016-01-19
中国药理学与毒理学杂志(2015年3期)2015-12-16
