
一测多评法测定肉苁蓉枳壳颗粒中7种成分的含量
2019-03-14 02:35黄乾峰李浩贤林华庆余楚钦
广东药科大学学报 2019年1期
黄乾峰,李浩贤,林华庆,余楚钦
(广东药科大学/广东省药物新剂型重点实验室,广东 广州 510006)
肉苁蓉枳壳颗粒处方由肉苁蓉、枳壳、白术组成。处方中肉苁蓉养血润肠,枳壳调畅气机,以助大肠推动之力,故可用于各种虚秘,尤其适用于老年性便秘[1]。松果菊苷、毛蕊花糖苷、管花苷A、异毛蕊花糖苷等苯乙醇苷类成分为肉苁蓉提取物中的主要活性成分[2],枳壳提取物的主要活性成分为柚皮苷、橙皮苷、新橙皮苷[3]。现行《中国药典》对枳壳项目下柚皮苷和新橙皮苷有定量要求[4]246,便通胶囊项下肉苁蓉也有定量要求,要求测定肉苁蓉中的松果菊苷[4]1253;其他活性成分研究文献也有相关的报道[5]。《中国药典》中主要以单一成分为指标的质量控制,无法完全反映中药“整体”的理念,而测定多指标的质量控制方法则需要对照品的种类和数量均比较大,且部分对照品价格昂贵(毛蕊花糖苷、松果菊苷等)。
一测多评法(QAMS)是在多指标质量控制时,以样品中对照品廉价易得的典型成分为内标,建立该成分与其他成分间的相对校正因子(RCF),再通过RCF计算出其他成分的量的方法,可实现使用少量对照品对多种成分的同步定量测定[6]。本研究采用QAMS法对肉苁蓉枳壳颗粒的7种有效成分进行同时测定,以成本低廉的柚皮苷为内参物,分别建立与松果菊苷、毛蕊花糖苷、管花苷A、异毛蕊花糖苷、橙皮苷、新橙皮苷的RCF,用外标法(ESM)测定柚皮苷的质量分数,根据RCF,计算其他成分的质量分数,并将QAMS的结果与外标法的结果进行比较,验证QAMS法结果的准确性和可靠性。
1 仪器与材料
1.1 仪器
高效液相色谱仪(美国Dionex公司);色谱柱为Jade-pak ODS-AQ(5 μm,250 mm×4.6 mm)、YMC-Pack Pro C18(5 μm,250 mm×4.6 mm)、Diamonsil C18(2)(5 μm,250 mm×4.6 mm);AJ0-4287 Security GuardTMCartridges C18保护柱;CP225D型电子分析天平(德国Sartorius公司);BS224S型电子分析天平(德国Sartorius公司);KH-400KDB型高功率数控超声清洗器(昆山超声仪器有限公司)。
1.2 药品与试剂
肉苁蓉枳壳颗粒(自制产品,批号20180908,20180913,20180917);松果菊苷(批号111670-200502、111670-201706,质量分数89.7%)、毛蕊花糖苷(批号111530-201713,质量分数92.5%)、柚皮苷(批号110722-201714,质量分数93.4%);橙皮苷(批号110721-201617,质量分数96.1%);新橙皮苷(批号111857-201703,质量分数99.2%)均由中国食品药品检定研究院提供;管花苷A(批号M0906AS,质量分数98%)、异毛蕊花糖苷(异类叶升麻苷,批号 M0106AS,质量分数98%)由大连美仑生物技术有限公司提供;甲醇(色谱纯);水(屈臣氏蒸馏水);甲酸(天津科密欧,批号 20180110,HPLC级)。
2 方法与结果
2.1 色谱条件
Jade-pak ODS-AQ 色谱柱(250 mm×4.6 mm,5 μm);流动相:甲醇-0.1%甲酸水溶液,梯度洗脱(0~20 min,30%~36.6%甲醇;20~25 min,36.6%~33.4%甲醇;25~45 min,33.4%甲醇;45~55 min,30%甲醇);流速:1.0 mL/min;检测波长:283 nm;柱温:35 ℃;进样量:10 μL。
2.2 溶液的制备
2.2.1 混合对照品溶液的制备 分别精密称取松果菊苷、毛蕊花糖苷、管花苷A、异毛蕊花糖苷、柚皮苷、新橙皮苷、橙皮苷适量,以50%(体积分数,下同)甲醇溶解并定容,制成质量浓度分别为松果菊苷0.202 mg/mL、毛蕊花糖苷0.204 mg/mL、管花苷0.204 mg/mL、异毛蕊花糖苷0.200 mg/mL、柚皮苷0.210 mg/mL、新橙皮苷0.202 mg/mL、橙皮苷0.400 mg/mL的混合对照品溶液。
2.2.2 供试品溶液的制备 取本品单一包装量,研细,取约1 g,精密称定,置具塞锥形瓶中,精密加入50%甲醇20 mL,称定质量,超声处理40 min,取出放冷后,称定质量,以50%甲醇补足减失的质量,摇匀,0.22 μm微孔滤膜滤过,即得。
2.2.3 阴性对照溶液的制备 取处方比例及同样制备工艺制得的不含肉苁蓉、枳壳的阴性对照样品,按供试品溶液的制备方法制成阴性对照溶液。
2.3 系统适用性及专属性试验
分别精密吸取混合对照品溶液、供试品溶液及阴性对照品溶液各10 μL进样,结果如图1。可见,松果菊苷、毛蕊花糖苷、管花苷A、异毛蕊花糖苷、柚皮苷、新橙皮苷、橙皮苷与其邻近的色谱法分离度皆大于1.5,拖尾因子在1.04~1.10之间,理论塔板数均在10 000以上,阴性对照在相应位置处未见色谱峰,方法专属性良好。
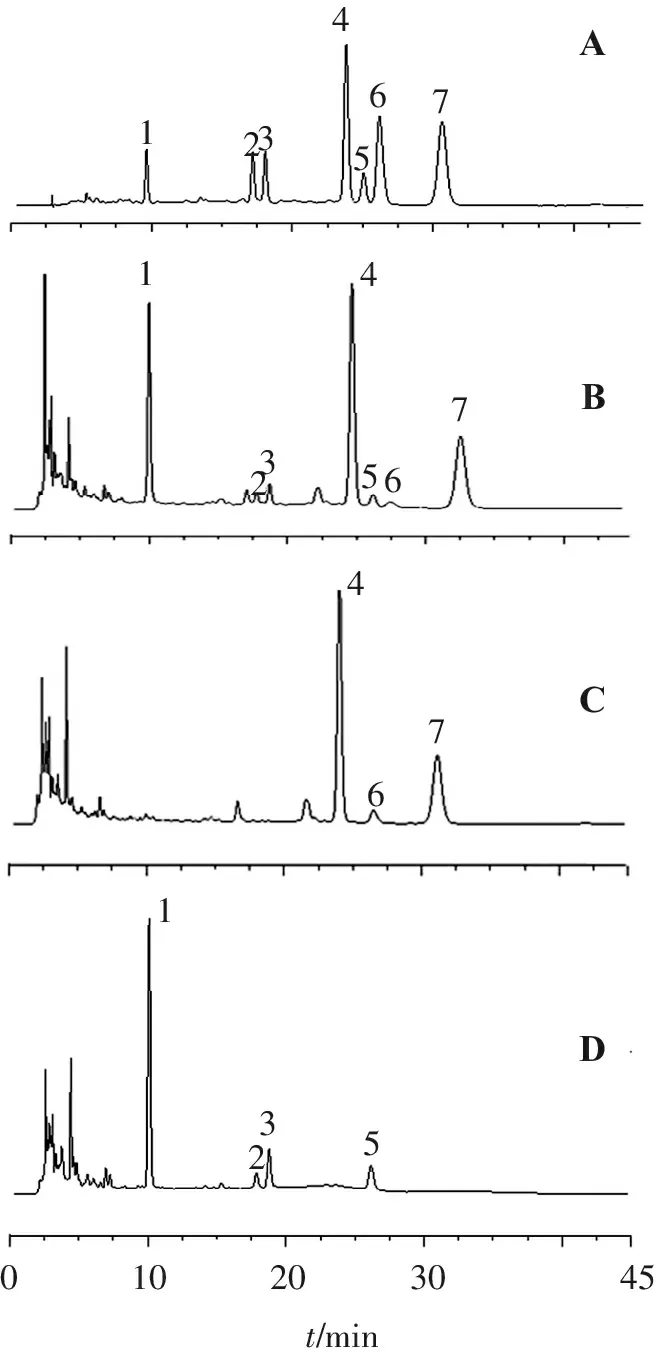
010203045t/minABCD543211674765327654321
A.混合对照品; B.供试品; C.阴性样品(缺肉苁蓉); D.阴性样品(缺枳壳); 1.松果菊苷; 2.管花苷A; 3.毛蕊花糖苷; 4.柚皮苷; 5.异毛蕊花糖苷; 6.橙皮苷; 7.新橙皮苷。
图1高效液相色谱图
Figure1HPLC chromatograms
2.4 线性关系考察
精密量取“2.2”项下混合对照品溶液适量进行稀释,得到系列混合对照品溶液,按“2.1”项下色谱条件分别进样检测,记录色谱图。以对照品质量浓度(x,μg/μL)为横坐标,峰面积(y)为纵坐标,绘制标准曲线,结果见表1。
2.5 稳定性试验
取同一供试品溶液,分别于制备后0、2、4、8、12 h进样10 μL测定,记录各组分色谱峰峰面积,计算RSD值。结果松果菊苷、毛蕊花糖苷、管花苷A、异毛蕊花糖苷、柚皮苷、新橙皮苷、橙皮苷的峰面积的RSD分别为0.75%、0.30%、0.34%、0.60%、0.84%、0.90%、0.43%,表明供试品溶液在12 h内稳定。
2.6 精密度试验
取同一供试品溶液,连续重复进样 6次,记录各组分色谱峰峰面积,计算 RSD值。结果松果菊苷、毛蕊花糖苷、管花苷A、异毛蕊花糖苷、柚皮苷、新橙皮苷、橙皮苷的峰面积的RSD分别为0.69%、1.69%、0.54%、1.34%、0.64%、1.80%、0.35%,表明仪器精密度良好。
2.7 重复性试验
精密称取同一供试品,按“2.3”项下平行制备供试品溶液 6 份,分别进样10 μL测定,记录各组分色谱峰峰面积,计算RSD值。结果松果菊苷、毛蕊花糖苷、管花苷A、异毛蕊花糖苷、柚皮苷、新橙皮苷、橙皮苷的平均含量(n=6)分别为4.312 1、5.312 1、0.891 2、0.468 9、0.461 2、1.085 6、2.300 2 μg/g,RSD分别为0.69%、1.79%、1.19%、1.43%、0.58%、1.70%、0.51%,表明方法重复性良好。
2.8 加样回收率试验
取已知质量分数的肉苁蓉枳壳颗粒,研细,取约1 g,精密称定6份,按照“2.2.2”项下方法制备溶液。精确量取1 mL至2 mL容量瓶内,再精确加入1 mL的松果菊苷、毛蕊花糖苷、管花苷A、异毛蕊花糖苷、柚皮苷、橙皮苷、新橙皮苷混合对照品溶液,混匀。依次取10 μL滤液进样测定。结果测得松果菊苷的平均回收率为100.8%(RSD=0.7%)、毛蕊花糖苷的平均回收率为99.50%(RSD=1.4%)、管花苷的平均回收率为101.1%(RSD=1.6%)、异毛蕊花糖苷的平均回收率为102.4%(RSD=0.60%)、柚皮苷的平均回收率为100.7%(RSD=1.2%)、新橙皮苷的平均回收率为101.4%(RSD=1.5%)、橙皮苷的平均回收率为 99.83%(RSD=1.1%)。

表1 7种成分的线性关系考察结果Table 1 Linear investigation result of 7 constituents
2.9 校正因子(fk/s)的计算
2.9.1 多点校正法 依照文献[7]方法,取6个质量浓度的混合对照品溶液分别测定,计算各浓度点所得的fk/s,取平均值作为定量用fk/s,结果见表2。校正因子(fk/s)计算公式:fk/s=(Cs×Ak)/(Ck×As);待测成分质量浓度计算公式:Ck′=(Cs×Ak′)/(fk/s×As),式中:Cs为参照物质量浓度,As为参照物色谱峰峰面积,Ck为其他对照组分质量浓度,Ak为其他对照组分色谱峰峰面积,Ck′为待测组分质量浓度,Ak′为待测组分色谱峰峰面积。
2.9.2 斜率校正法 在标准曲线Y=aX+b中,X=(Y-b)/a=Y/a-b/a,由于b值通常为误差引起,在a/b值大于100时,b/a值可以忽略不计,此时可以X=Y/a直接计算。故fk/s可以二者的斜率a之比直接计算,即fk/s=ak/as,即可以参照物快速推算其余待测成分的量Ck′[Ck′=Ak′/(as×fk/s)],式中:as为参照物斜率,ak为其他对照组分斜率。应用此法需先建立参照物的标准曲线获得其as。本文以斜率校正法测得松果菊苷、管花苷A、毛蕊花糖苷、异毛蕊花糖苷、橙皮苷、新橙皮苷相对于柚皮苷的fk/s分别为0.379、0.422、0.563、0.600、0.453、1.14。
2.9.3fk/s的耐用性考察 考察了不同色谱柱、不同流速、不同柱温的重现性,考察其耐用性,结果见表3。结果显示,校正因子的耐用性良好。
2.9.4 待测组分色谱峰的定位 柚皮苷通过对照品定位,其他待测组分采用相对柚皮苷保留时间(relative retention time;RRT值)进行定位。采用一测多评法进行测定时,通过柚皮苷的保留时间计算其他待测组分峰的RRT数值,根据RRT值及光谱吸收可对其他6个组分进行准确定位。本实验测定了相对保留值在不同仪器、不同色谱柱、不同流速、不同柱温的重现性,结果见表4。可见,各待测组分与柚皮苷的相对保留时间RSD均<5%。
2.10 QAMS法与外标法测定结果的比较
取3个不同批次的样品分别进样,先用外标法对肉苁蓉枳壳颗粒中7种不同成分进行测定,再用QAMS法建立待测成分间的RCF对其他成分的质量分数进行计算,比较2种方法的计算结果,结果见表5。可见,2种方法所测得结果差异无统计学意义(相对误差均<5%),表明建立的QAMS法可用于肉苁蓉枳壳颗粒的多成分质量评价研究。

表2 以柚皮苷为参照的fk/s(多点校正法)Table 2 fk/s with naringin as reference (multi-point correction method)

表3 校正因子(fk/s)的耐用性试验结果Table 3 Robustness test of correction factors
注:3根色谱柱在检测时带AJ0-4287 SecurityGuardTMCartridges C18保护柱。

表4 不同条件下柚皮苷与其他组分的相对保留时间Table 4 Relative retention time between naringin and other components under different conditions
表5 QAMS法与外标法测定肉苁蓉枳壳颗粒中7种成分的质量分数的结果比较
Table5Determination of seven components in Cistanche- Fructus aurantii granules by QAMS and external standard method(n=6)w/(μg·g-1)

3 讨论
参考《中国药典》中“肉苁蓉”项下的前处理方法[4] 135-136,提取溶剂考察了体积分数分别为40%、50%、60%、70%的甲醇溶液,结果显示50%甲醇提取时能将各组分提取完全;同时,考察了提取溶剂用量以及超声时间的影响,结果表明加入液料比1∶20(g∶mL)的50%甲醇超声处理40 min即可提取完全。
参考《中国药典》中“便通胶囊”项下色谱条件[4]1253,考察了甲醇-水、甲醇-0.1%甲酸水、甲醇-0.5%甲酸水溶液系统,结果显示以甲醇-0.1%甲酸水系统的峰型更好、分离效果良好,本文选用甲醇-0.1%甲酸水系统作为流动相。
在待测的7个成分中,以柚皮苷的质量分数较高、且相对独立,对照品价格便宜以及化学性质稳定,所以选用柚皮苷作为内参物。
本研究初步建立了一测多评法用于本研究所开发的肉苁蓉枳壳颗粒中的7种成分的同步测定,与外标法实测值没有显著差异(相对偏差<5%),表明建立的方法具有较好的可信度。本法与外标法相比,能极大地节约多成分质量分数测定的检测成本,并为肉苁蓉枳壳颗粒药材、提取物、制剂的多指标质量控制模式提供新方法,为质量标准完善和质量评价提供参考依据。
猜你喜欢
中西医结合心脑血管病杂志(2022年20期)2022-11-08
中国中医药现代远程教育(2022年17期)2022-09-26
世界科学技术-中医药现代化(2021年10期)2021-03-02
中成药(2020年8期)2020-09-15
天然产物研究与开发(2018年2期)2018-04-04
益寿宝典(2017年2期)2017-02-26
恋爱婚姻家庭·养生版(2017年2期)2017-02-15
甘肃林业(2016年2期)2016-11-07
中国当代医药(2015年24期)2015-03-01
中国当代医药(2015年9期)2015-03-01
