
基于线粒体Cytb序列的伯氏肩孔南极鱼群体遗传结构初步分析
2019-03-08 02:49:46马春艳冯春雷王鲁民张凤英蒋科技赵宪勇马凌波
海洋渔业 2019年1期
赵 娜,马春艳,宋 炜,冯春雷,王鲁民,张凤英,蒋科技,赵宪勇,马凌波
(1. 中国水产科学研究院东海水产研究所,上海 200090; 2. 上海海洋大学,上海 200090;3. 中国水产科学研究院黄海水产研究所,青岛 266071)
在大约5亿9千万年前的前寒武季后期,超级大陆冈瓦纳还包括南极洲、非洲、南美洲、澳大利亚、印度和阿拉伯半岛[1]。自中生代晚期,剧烈的大陆板块活动使得超级大陆冈瓦纳开始逐渐解体,几个大洲开始分离开来,到了新生代时期南极大陆经过缓慢的漂移,逐渐到了现在的地理位置[2]。在南极大陆逐渐漂移的过程中,整个南极大陆的气候开始逐渐变冷,慢慢被冰雪覆盖[3-4]。南大洋的水温在南极大陆逐渐漂移过程中,水温不断降低,许多生物走向灭绝,仅有少数能够适应低温的生物存活了下来,南极鱼类为了适应低温环境,种群进行了相应的改变,生物多样性也发生了改变。南极大陆被南大洋分割开来,环绕南极的南极绕极流( Antarctic Circumpolar Current, ACC),阻碍了南极海域与其它海域的联系,对南极鱼类的种群遗传具有一定的影响。
南极鱼类在始新世到现在的4000万年内发生了显著性的变化[5]。虽然南大洋仅仅占了全世界海洋面积的10%,现有的鱼类有322种[6]。伯氏肩孔南极鱼(Trematomusbernacchii)属于硬骨鱼纲(Osteichthyes),鲈形目(Perciformes),南极鱼亚目(Notothenioide),南极鱼科(Notothenioidei),肩孔南极鱼属,是一种广泛分布于南极附近的适应寒冷环境的鱼类,因其在寒冷条件下的适应性机制和自适应辐射具有重要的科学研究价值以及其经济价值而引起了世界范围的关注[7]。目前,关于伯氏肩孔南极鱼遗传多样性、遗传分化和种群历史动态等方面的研究报道较少。本研究采用mtDNA细胞色素b(Cytb)基因序列的分析技术,研究分析了南极地区伯氏肩孔南极鱼的种群遗传结构及其多样性与种群历史动态,以期为更好地保护该鱼种野生种群、促进其资源的可持续利用提供科学依据。
1 材料与方法
1.1 实验材料
实验所用的伯氏肩孔南极鱼分别采自位于罗斯海(LSH)、凯西站(KXZ)、长城站(CCZ)、戴维斯站(DWSZ)和中山站(ZSZ)的5个采样点(表1)。样品采集的时间、地点以及数量见图1与表1。样品采集后先对样品进行形态鉴定,在-20 ℃条件下冷冻保存运回到实验室。

表1 伯氏肩孔南极鱼的采集样品数Tab.1 Sample number of Trematomus bernacchii in each location
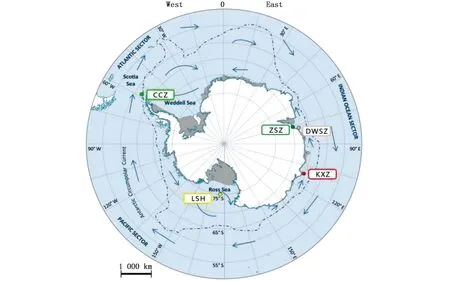
图1 伯氏肩孔南极鱼取样地点图Fig.1 Sampling sites of Trematomus bernacchii注:CCZ:长城站,LSH:罗斯海,KXZ:凯西站,DWSZ:戴维斯站,ZSZ:中山站Note:CCZ:Great Wall Station,LSH:Ross Sea,KXZ:Casey Station,DWSZ:Davis station,ZSZ:Zhongshan Station
1.2 伯氏肩孔南极鱼mtDNA Cytb 序列的扩增与测序
取伯氏肩孔南极鱼背部肌肉或鳍部约100 mg,使用海洋动物组织基因组DNA提取试剂盒(北京,天根生化科技有限公司)提取总DNA。
1)Cytb引物使用自行设计的引物如下所示:
CF1/R1:CCGAAACTGTCCTTGGTGTGAG、GAGTGAGGGTGGCGTTATCTAC
CF2/R2:ACCTCCCCGCTCCATCTAAT、TACCCCCCCAAGTTTGTCTG
CF3/R3:AGGGGGGTTTTCGGTAGATA、TTTAGGGTCCTGGGGGCTTC
2)PCR在25μL的反应体系中执行,其中包括:
2xTaqPCR Master-Mix 12.5 μL
上游引物(10mmol·L-1) 1 μL
下游引物(10mmol·L-1) 1 μL
模板DNA 1 μL
去离子水 9.5 μL
3)PCR反应条件为:
预变性 94℃ 5min;
变性 94℃ 30s
退火 54℃ 45s
延伸 72℃ 1min;
第二阶段循环35次
总延伸 72℃ 7min
PCR产物经电泳检测质量和大小后,由JIE LI BILOGY进行正向测序。
1.3 数据分析
利用Clustal X[8]对序列进行对位排列, 并结合人工核查与校正。单倍型数目及分布、核苷酸多样性使用DnaSP 5.1[9]软件确定。用MEGA 5.1[10]软件按照Kimura-2-parameter[11]进化模型来构建单倍型邻接关系树, 系统树的可靠性采用1000次重复抽样评估,并计算群体内的遗传距离。
使用Arlequin 3.1[12]软件的分子变异分析[13](AMOVA)来评估群体间遗传变异,通过1000次重复抽样来检验不同遗传结构水平上的协方差的显著性,按样本的地理来源将5个群体分为1个组群,并采用Exact检验检测单倍型在两两群体间分布频率的差异来评价群体间的遗传分化[14]。上述分析均由Arlequin 3.1软件计算,群体间的遗传距离采用Kimura-2-parameter模型计算。
2 结果与分析
2.1 序列变异特征分析
本实验共得到了5个群体98尾伯氏肩孔南极鱼的线粒体,Cytb基因全序列长度为1 141 bp,无碱基的插入与缺失,转换替代明显多于颠换,大多数变异发生在密码子第3位(79.73%),第1位(10.81%)和第2位(9.46%)的变异较低。其突变位点为75个,变异比率为6.57%,其中单一信息位点47个,占4.12%,简约信息位点28个,占2.45%。4个碱基A、T、C和G的平均频率分别为20.6%、32.4%、26.7%和20.3%,其中G的碱基含量低于其它3种碱基含量,A+T(53%)的出现频率高于C+G(47%),表现出了其碱基组成具有偏向性。
2.2 单倍型在群体中的分布及遗传关系
98个个体中共检测到27种单倍型,所得单倍型已全部提交到GenBank,登录号为(MF664438~MF664464 )。伯氏肩孔南极鱼27种单倍型在5群体中的分布如表2所示。群体间共享单倍型4个,占单倍型总数的14.81%,剩余23个均为某个群体所特有,在共享单倍型中Hap2为5个群体所共有,Hap4为4个群体所共有(除罗斯海外),Hap7为凯西站和戴维斯站所共有,Hap23为凯西站和中山站所共有,各个站点存在共享单倍型,说明其亲缘关系较近,各地理种群具有一定的基因交流,是一个随机交配的群体。伯氏肩孔南极鱼5个群体的遗传多样性数据见表3,98尾伯氏肩孔南极鱼的单倍型多样性为0.685 90±0.002 36,核苷酸多样性为0.002 59±0.013 43,遗传多样性较高。核苷酸多样性分析表明(表3)中山站核苷酸遗传多样性较高,大于0.004。
2.3 种群遗传距离与遗传分化
从5个伯氏肩孔南极鱼地理群体的遗传距离(表4)来看,群体内的平均遗传距离在0.001 254~0.010 199,群体间的平均遗传距离在0.001 733~0.007 140。结果表明,5个群体间的平均遗传距离与群体内的平均遗传距离处于同一水平。
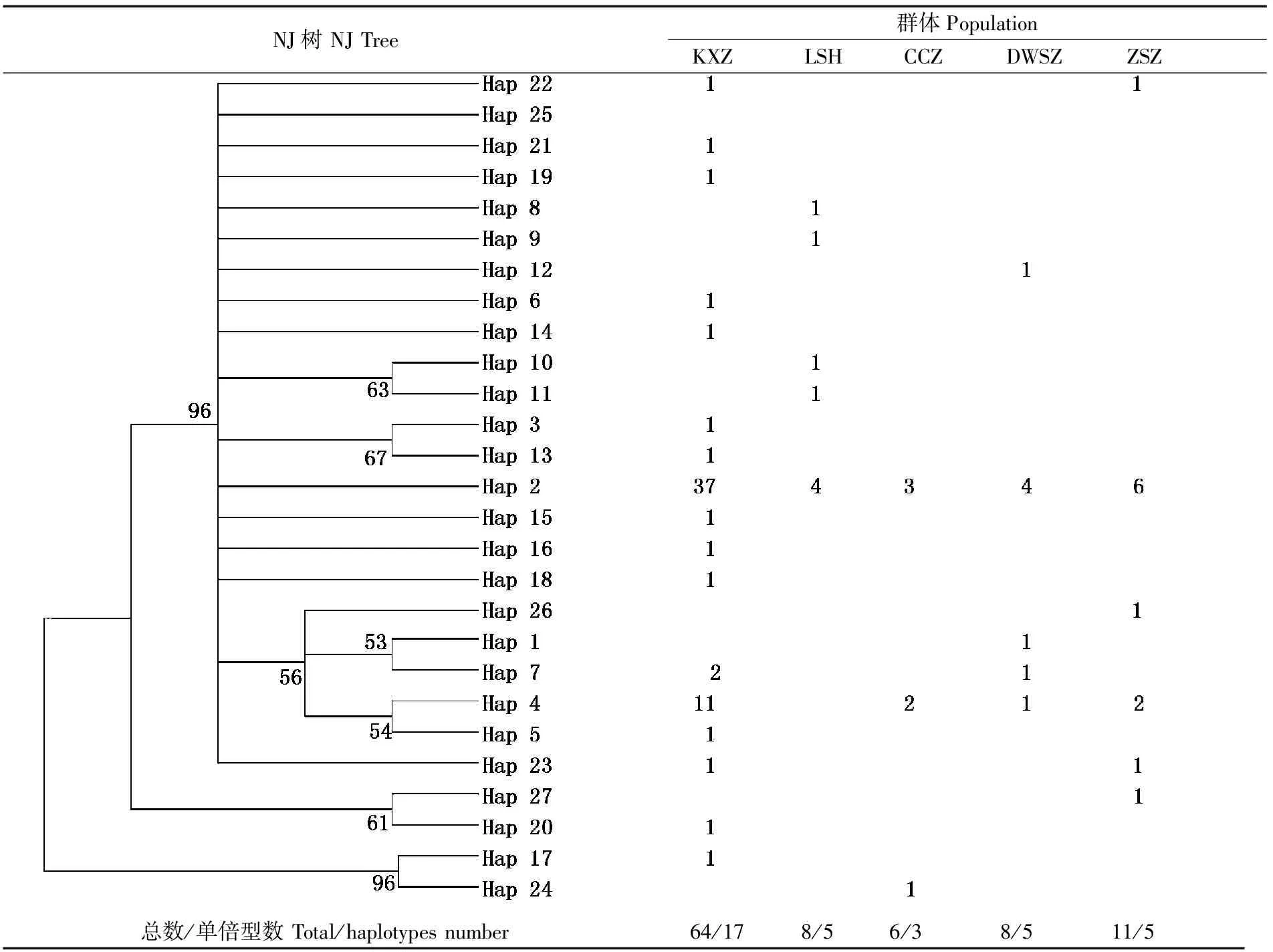
表2 伯氏肩孔南极鱼27种单倍型在5个群体中的分布及其分子系统树Tab.2 Distribution of 27 haplotypes among 5 Trematomus bernacchii populations and its NJ molecular phylogenetic tree
注:节点数字表示大于50%的Bootstrap支持率;CCZ:长城站,LSH:罗斯海,KXZ:凯西站,DWSZ:戴维斯站,ZSZ:中山站
Note:Numbers at nodes indicate bootstrap values greater than 50% with 1000 replicates;CCZ:Great Wall Station,LSH:Ross Sea,KXZ:Casey Station,DWSZ:Davis Station,ZSZ:Zhongshan Station

表3 不同群体伯氏肩孔南极鱼的遗传多样性参数Tab.3 Parameters of genetic diversity of different populations of Trematomus bernacchii
注:CCZ:长城站,LSH:罗斯海,KXZ:凯西站,DWSZ:戴维斯站,ZSZ:中山站
Note:CCZ:Great Wall Station,LSH:Ross Sea,KXZ:Casey Station,DWSZ:Davis station,ZSZ:Zhongshan Station
在群体遗传学中两两之间的遗传分化指数一般分布在-1~1之间,遗传分化系数(Fst)越大表示群体间的分化程度越高[15]。伯氏肩孔南极鱼的5个群体间的Fst在-0.002 450~ 0.190 380之间(表4),5个群体间的遗传分化水平较低(Fst= -0.018 300~0.190 380,P>0.05)。AMOVA分析表明大部分的遗传变异来自于种群内部,只有少数遗传变异来自于种群之间,并且Φstatistics为正值。Exact检验表明,单倍型在群体间分布频率的差异显著(P=0.001 96),说明其种群之间存在基因交流。
2.4 中性进化分析
5个伯氏肩孔南极鱼群体的中性检验分析结果见表3。Tajima’s D检验的结果表明除戴维斯站(DWSZ)外均为负值,凯西站(KXZ)为极显著差异(P<0.01),中山站(ZSZ)显著性差异(P<0.05),其它群体均为不显著差异(P>0.05)。中山站(ZSZ)显著的偏离突变-漂变平衡(P<0.05),凯西站(KXZ)极显著的偏离突变-漂变平衡(P<0.01)。
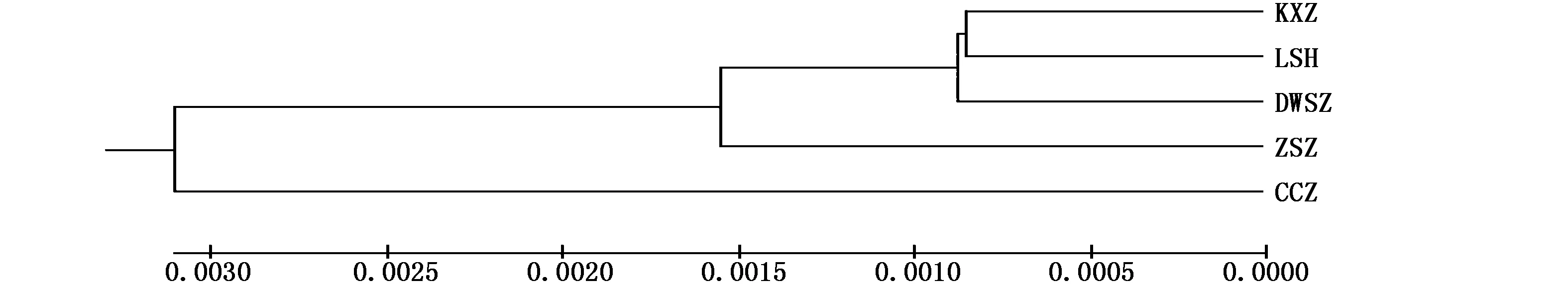
图2 基于K-2-P遗传距离的伯氏肩孔南极鱼5个群体之间的UPGMA树Fig.2 UPGMA tree of 5 Trematomus bernacchii populations built under K-2-P genentic distance注:CCZ:长城站,LSH:罗斯海,KXZ:凯西站,DWSZ:戴维斯站,ZSZ:中山站Note:CCZ:Great Wall Station,LSH:Ross Sea,KXZ:Casey Station,DWSZ:Davis Station,ZSZ:Zhongshan Station

群体PopulationKXZLSHCCZDWSZZSZKXZ 0.001 9800.001 830 0.001 733 0.003 104 0.005 875LSH0.039 640 0.001 254 0.001 7350.003 221 0.005 912CCZ0.190 380 0.097 110 0.010 1990.003 031 0.005 851DWSZ -0.002 450 0.156 2800.071 790 0.001 6590.007 140ZSZ0.011 1500.011 940 0.013 940-0.018 300 0.004 584
注:CCZ:长城站,LSH:罗斯海,KXZ:凯西站,DWSZ:戴维斯站,ZSZ:中山站
Note:CCZ:Great Wall Station,LSH:Ross Sea,KXZ:Casey Station,DWSZ:Davis Station,ZSZ:Zhongshan Station
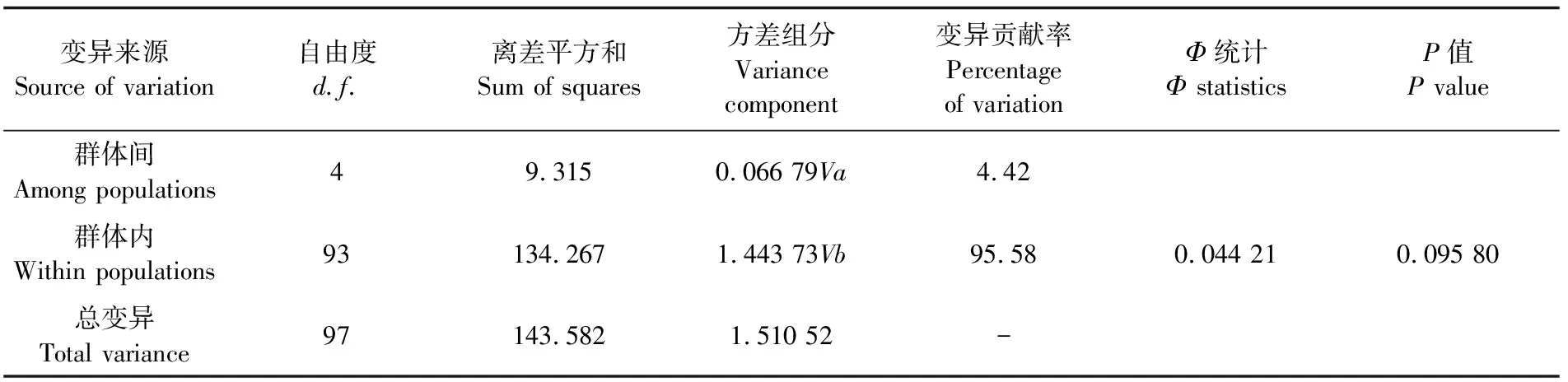
表5 伯氏肩孔南极鱼群体遗传差异的 AMOVA 分析Tab.5 Analysis of molecular variance (AMOVA) among populations of Trematomus bernacchii
Fu’s 检验凯西站(KXZ)、罗斯海(LSH)和戴维斯站(DWSZ)群体为正值,长城站(CCZ)和中山站(ZSZ)群体为负值,且除凯西站(KXZ)外其余均无显著性差异(P>0.05),Fs值的显著性表明凯西站极显著的偏离突变-漂变平衡(P<0.01),Fs是负值,并且显著的偏离中性(P<0.05),则有可能是群体扩张导致的。伯氏肩孔南极鱼群体的D值和Fs均为负值,且具有极显著的差异(P<0.01),因此可以推断出其可能经历了种群的快速爆发与扩张事件。
2.5 历史统计分析
将本实验中的5个群体进行错配分析,得到不同的频率分布形状图,展现了伯氏肩孔南极鱼群体的种群历史多样性,其中SSD和Raggedness值较大且不存在显著差异(P<0.05)。群体扩张参数τ为1.723 74,参考线粒体Cytb的进化速率为每百万年0.9%~1.2%,伯氏肩孔南极鱼的性成熟年龄为8年[16],根据公式T=τ/2u,得到该种群的扩张时间大约为9.84-10.49万年前,大约是中更新世时期,该时期气候寒冷,有冰期与间冰期的明显交替[17]。
3 讨论
3.1 遗传多样性
通过研究Cytb的完整序列基因分析了南极地区5个伯式肩孔南极鱼地理群体的遗传多样性,研究结果以在一定的程度上反映伯氏肩孔南极鱼资源的遗传背景。有些群体数量较少,主要是由于南极鱼类比较难获取样本,统计分析表明用于群体分析数量最少为5个,5个样本即可以对数据进行溯祖分析[18]。测序后得到的序列长度为1 141 bp,无碱基的插入与缺失,转换替代明显多于颠换,大多数变异发生在密码子第3位(79.73%)。整个群体单倍型多样性较高(H=0.685 90±0.002 36)、核苷酸多样性较低(Pi=0.002 59±0.013 43)的模式,这种模式揭示其出现过种群瓶颈效应[19],这与在银鲳(Pampusargenteus)[20]、松江鲈(Trachidermusfasciatus)[21]、小黄鱼(Larimichthyspolyactis)[22]等研究中得到的结果相似。相比同生活在南大洋的物种来说,如南极东部沿岸贝氏肩孔南极鱼(H=0.994,Pi=0.012 71)及尼氏肩孔南极鱼(H=0.978,Pi=0.004 58),都与伯氏肩孔南极鱼群体有着相似的遗传多样性。贝氏肩孔南极鱼和尼氏肩孔南极鱼都属于GRANT等[19]划分的第二类遗传模式,而高H低Pi的现象是由于有效种群数量较少,此后经历一个快速的扩张期,积累了较多的变异,然而仍未积累大量序列变异造成[23]。在冰期鱼类可能面临恶劣的环境导致种群数量下降经历瓶颈期,冰期之后由于环境适宜,种群呈现爆炸式扩散,快速积累遗传变异,因此伯氏肩孔南极鱼是一个大的随机交流的群体。

图3 不同站点的伯氏肩孔南极鱼群体之间的错配分析Fig.3 Mismatch distribution of populations of Trematomus bernacchii from different sites注:a,凯西站;b,罗丝海;c,长城站;d,戴维斯站;e,中山站Note:a,Casey Station;b,Ross Sea:c,Great Wall Station;d,Davis Station;e,Zhongshan Station
3.2 遗传结构与分化
从鱼类对环境的适应能力、对生态的适应性进化能力以及对自然选择的效应等方面能分析出种群的遗传结构[24]。伯氏肩孔南极鱼的NJ树显示,该种群是一个随机交配的群体,5个群体间的遗传分化水平较低(Fst= -0.018 30~0.190 38,P>0.05),不存在显著的遗传分化现象,遗传距离相近,即不存在地理分布的谱系,这与UPGMA树得到的结构一致并与大多数的海洋性鱼类在非常广泛的地理范围内不存在显著的遗传差异的结果一致[19,25]。产生此现象的一个主要原因可能是由于南大洋存在南极绕极流,有助于伯氏肩孔南极鱼鱼卵的扩散,各个群体之间可以进行交流。鱼类在属、种和种群3级水平上的遗传距离值分别为0.9、0.3和0.05[26]。5个群体之间的遗传距离在0.001 252~0.009 991之间,均小于0.05,表明其采样点之间的分化尚未达到种群水平。AMOVA分析的结果也同样显示了变异主要来自种群内部,而整个大的群体中不存在分化现象。
3.3 种群的历史动态分析
种群在历史上所经历的变化主要是通过单倍型多样性与核苷酸多样性来推测,根据GRANT等[19]提出的历史事件的4个假设来看,伯氏肩孔南极鱼线粒体Cytb基因属于第2种类型,这一类型的种群在历史过程中发生过瓶颈效应或者奠基者效应,在此之后由一个小的群体迅速扩张与爆发。Tajima’sD和Fu’s检验结果显示在凯西站出现了极显著差异(P<0.01),其余群体无显著性差异(P>0.05),这可能与样本本身较难获取,数量上有所差异,导致结果存在差别。5个群体除罗斯海外其余采样点在错配分析图中均有小的峰值出现,可能预示了在这些地区存在一个比较年轻的祖先系统,值得注意的是罗斯海这一采样点无此现象,可能是由于除了本身环境中的南极绕极流的存在,该地区的洋流还存在自身的漩涡现象,还需要进一步的实地考察。根据公式得到该种群的扩张时间大约为9.84~10.49万年前,大约是中更新世时期,该时期气候寒冷,有冰期与间冰期的明显交替,气候条件的变化使整个群体经历瓶颈效应后扩张群体存活下来。
Exact检验结果表明了5个群体之间无显著的遗传分化,是一个可以随机交配的群体,但是Cytb完整序列分析结果显示不能简单地把南极地区作为整个单一种群进行管理,必须结合其它分子标记技术如SSR、AFLP、SNP等全面检测伯氏肩孔南极鱼群体的遗传结构,从而为渔业管理措施的制订提供支撑。
伯氏肩孔南极鱼分布较为广泛,本研究由于样本数量的问题及遗传背景资料的缺乏,结果尚不能全面反映伯氏肩孔南极鱼的遗传特征,今后研究中需要增加样本数量,并结合核基因标记联合分析伯氏肩孔南极鱼的种群遗传与分化。
猜你喜欢
成都信息工程大学学报(2022年4期)2022-11-18 07:32:34
故事家·花开不败(2019年11期)2019-09-10 17:16:30
青少年科技博览(中学版)(2018年6期)2018-08-16 06:51:10
发明与创新·大科技(2018年3期)2018-07-27 08:47:38
百科探秘·航空航天(2017年12期)2018-01-31 02:31:30
今日华人(2018年2期)2018-01-16 18:23:33
海洋世界(2016年3期)2016-03-24 08:43:05
儿童故事画报·自然探秘(2016年2期)2016-03-15 05:56:32
中国海洋大学学报(社会科学版)(2014年6期)2014-08-18 02:58:30
青年文摘·上半月(1986年6期)1986-11-01 04:17:32
