
戈谢病Ⅱ型1例报道并文献复习
2019-03-04 08:24史惠陈日玲
中国中西医结合儿科学 2019年1期
史惠, 陈日玲
戈谢病又称葡萄糖脑苷脂病,是一种罕见的常染色体隐性遗传病,其遗传方式符合经典的孟德尔理论。戈谢病是溶酶体贮积症中最常见的一种,由于溶酶体内葡萄糖脑苷酯酶(glucocerebrosidase,GBA)缺乏,导致大量的葡萄糖脑苷脂在肝、脾、骨骼、肺及脑组织的单核-巨噬细胞中贮积[1]。该病可累及多个系统,临床表现无特异性,极易漏诊或误诊[2]。广东医科大学附属医院近期接诊1例戈谢病患儿,现回顾性分析该患儿的临床资料,以提高儿科医师对该疾病的认识。
1 临床资料
患儿,女,7个月,主因痰鸣3月余,加重伴咳嗽2周,发热2 d入院。患儿系G2P2,孕39周顺产出生,出生时无窒息抢救史,生后无抽搐病史。生长发育较同龄儿落后,2个月开始抬头,3个月抬头不稳,5个月会翻身,现7个月,不会独坐,无追人追物反应。自5月龄开始易呛奶。父母及哥哥体健,父母非近亲结婚,家族中无类似病史。入院查体:体温38.3 ℃,脉搏185次/分,呼吸50次/分,体质量6.5 kg,身长65 cm,头围41.5 cm;急性面容,神志清,精神状态一般,颈强直,角弓反张,前囟平软,大小约1 cm×0.5 cm,双侧瞳孔等大等圆,对光反射灵敏,双眼凝视,眼球无水平及垂直运动,牙关紧闭,口唇轻度发绀,咽稍红,呼吸稍促,可见三凹征,以胸骨上窝明显,双肺呼吸音粗,可闻及大量痰鸣音及细湿啰音。心率185次/分,律齐,未闻及杂音。腹膨隆、柔软,未见腹壁静脉显露,肝脏肋下6 cm触及,质韧,脾脏肋下9 cm触及,质韧,肠鸣音正常,4次/分。四肢活动自如,肌力未见异常,肌张力增高,布氏征、双侧巴氏征及克氏征均阴性。
辅助检查:血常规三系正常,肝功能、肾功能、电解质、心肌酶、凝血功能、血氨、免疫球蛋白、T细胞绝对值计数、优生优育病毒抗体系列均未见异常。C反应蛋白9.28 mg/L;降钙素原0.51 μg/L。痰涂片:找到极少量革兰阴性杆菌及少量革兰阳性球菌,找到极少量酵母样孢子体,未找到抗酸杆菌;肺泡灌洗液涂片:找到极少量革兰阴性杆菌及少量革兰阳性球菌,未找到酵母样孢子体,未找到抗酸杆菌。胸部X线结果考虑支气管肺炎可能。双侧腕关节X线示骨质结构未见明显异常。纤维支气管镜下可见支气管内膜炎症及会厌软骨软化;行肺泡灌洗液细胞学检查,结果提示细胞总数(不含红细胞)约5.76×109/mL;肺泡巨噬细胞9%;中性粒细胞91%;外观无色浑浊,易见细菌,未见真菌及嗜酸性粒细胞;脂肪染色阴性。痰培养(肺泡灌洗液)可见铜绿假单胞菌,对美罗培南、亚胺培南、左氧氟沙星、头孢吡肟、庆大霉素、头孢他啶等抗生素敏感。心脏及胃肠道彩超未见异常;腹部彩超提示肝、脾大(均平脐水平);泌尿系彩超可见左肾结石(约0.3 cm×0.3 cm);颅脑彩超提示第三脑室稍扩张(0.44 cm),双侧大脑中动脉血流动力学指标未见明显异常。动态脑电图提示背景慢化。吞咽功能评估发现患儿基本无吸吮动作。骨髓涂片示骨髓三系增生活跃,形态无明显异常,可见稍多疑似戈谢细胞。住院第15天患儿开始出现抽搐,表现为四肢强直伴抖动、双眼向上凝视、牙关紧闭,口唇发绀伴心率、血氧下降。入院后予低流量吸氧、抗感染、化痰、补充维生素AD、止惊及营养支持等处理,患儿体温好转,肺部症状无明显改善,神经系统症状呈进行性加重。住院23 d后患者家属要求转院治疗,院外随访3 d后患儿死亡。
基因检测:因疑似戈谢病,为进一步明确诊断,经患儿家长知情同意,采集患儿及其父母的外周静脉血2 mL,乙二胺四乙酸管抗凝,应用基因测序及多重连接探针扩增技术(multiples ligation-dependent probe amplification,MLPA)进行溶酶体贮积症相关基因检测。结果显示,患儿GBA基因Exon11存在错义突变c.1448T>C(p.Leu483Pro,图2),预计会使所编码蛋白质的483位氨基酸残基由Leu变为Pro,通过家系来源验证,该突变遗传自其父亲母亲。
2 讨论
戈谢病是一种世界范围性罕见病,1882年由法国医师Philippe Gaucher首次报道而得名[3]。其发病率在不同种族及地域间差异较大,据报道,全球戈谢病发病率很低,1∶50 000~1∶40 000,而在德国犹太人中发病率可高达1∶855,我国自1948年首次报道以来,各地区均有报道,总体发病率1∶200 000~1∶500 000[4-5]。
根据有无神经系统受累及起病急缓,临床上将戈谢病分为3型:Ⅰ型(慢性型)、Ⅱ型(急性神经型)和Ⅲ型(亚急性神经型)[6]。临床表现与GBA缺乏的程度相关。其典型临床表现有:(1)肝脾肿大,尤以脾肿大明显,继发脾功能亢进后可有贫血、血小板减少等;(2)骨骼改变,包括骨密度降低、病理性骨折等,X线可见烧瓶样骨破坏;(3)生长发育落后甚至倒退;(4)肺部受累出现咳嗽、呼吸窘迫;(5)神经系统受累,出现颈强直、角弓反张、牙关紧闭、肌张力增高、吞咽功能障碍、眼球运动失调、惊厥发作等[2]。本例患儿以严重肺部感染为首发症状就诊,入院查体除肺部体征外,有颈强直、角弓反张、眼球运动失调、四肢肌张力增高,吸吮、吞咽功能不协调等中枢神经系统受累表现,可触及明显肝脾肿大,以脾为甚,考虑为戈谢病Ⅱ型。
戈谢病的致病基因即编码GBA基因定位于人类染色体1q21,基因全长包含2 564个碱基,有11个外显子,10个内含子。GBA基因的突变形式多种多样,包括错义突变、无义突变、大片段缺失或插入、基因与假基因融合、基因转化及剪切位点突变等,其中约76%为错义突变[7]。目前国际上已报道的基因变异位点达500余种[8],不同种族间有明显差异,德裔犹太人以N370S突变最为常见,我国则以L444P最为常见[9]。另外,基因突变类型与临床表型的关系尚不确定,据文献报道,L444P可见于各型戈谢病患者,纯合子大多表现为亚急性神经病变型(Ⅲ型),而c.1342G>C(D409H)纯合子通常表现为心血管型[10]。当临床有不明原因肝脾肿大合并神经系统症状时,要考虑到戈谢病可能。目前,戈谢病诊断的金标准是β-葡萄糖脑苷脂酶活性检测,患者外周白细胞或皮肤成纤维细胞中GBA活性降低至正常值的30%以下时,可确诊为戈谢病。血浆壳三糖酶活性检测[12]、骨髓形态学检查均可作为辅助诊断依据,而基因检测则对实验室要求较高,既可作为诊断依据,同时也可以指导治疗。
过去戈谢病的治疗主要以支持疗法为主,贫血者予输血治疗,脾亢者予切脾治疗等。近20余年来经过反复的临床实践,酶替代疗法已得到广泛认可。伊米苷酶是目前国内唯一可获得批准的戈谢病特异性治疗药物,可显著改善Ⅰ型戈谢病患者的临床症状和体征,提高患者生活质量,但对于Ⅱ型和Ⅲ型患者尚无有效的治疗方法[13]。段彦龙等[14]曾对北京儿童医院诊断的72例戈谢病患儿进行伊米苷酶替代治疗,结果提示可显著改善贫血、血小板减少,肝脾肿大、骨痛等症状。其他治疗如葡糖脑苷脂酶抑制
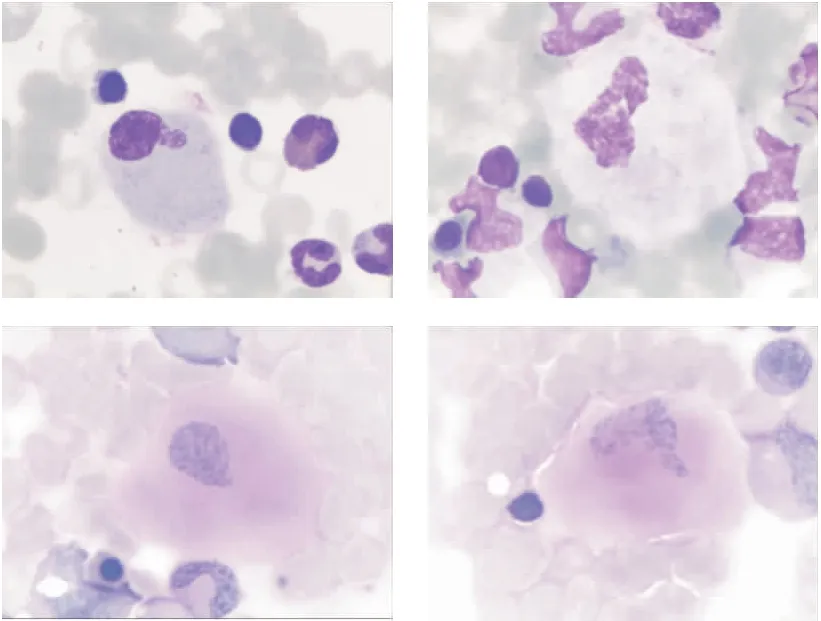
图1 患儿骨髓象(瑞氏染色及糖原染色×1 000)
剂在国外已被批准用于成人戈谢病,而基因治疗、造血干细胞移植等尚处于实验研究阶段[15-16]。该患儿由于起病早,病情进展快,尚未得到及时治疗即死亡。
综上所述,戈谢病作为少数可以被治疗的罕见病,早诊断、早治疗对于有效延缓疾病的进展、改善临床症状有着重要意义。如患儿有肝脾肿大、生长发育迟缓及神经系统受损表现,临床儿科医师应提高警惕,注意戈谢病可能,尽早完善家系基因学检查进行基因诊断或酶学检测进行确诊。对于已生育过戈谢病患儿的家庭及亲属,应进行遗传咨询及致病基因检测,若再次妊娠,应进行产前基因诊断避免患儿出生。
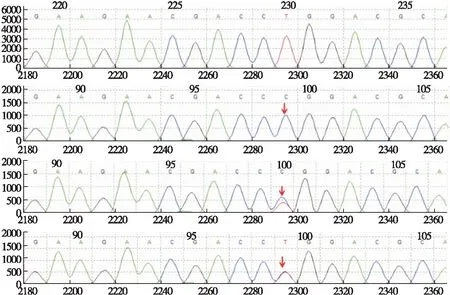
图2 患儿基因测序图
猜你喜欢
现代仪器与医疗(2022年3期)2022-08-12
中国典型病例大全(2022年13期)2022-05-10
中国典型病例大全(2022年7期)2022-04-22
昆明医科大学学报(2021年12期)2021-12-30
疯狂英语·新读写(2021年10期)2021-12-07
天津医科大学学报(2021年4期)2021-08-21
中华养生保健(2020年5期)2020-11-16
奥秘(2019年8期)2019-08-28
商周刊(2017年7期)2017-08-22
小猕猴智力画刊(2016年6期)2016-05-14
