
Uncovering the immunological repertoire of the carpet shell clam Ruditapes decussatus through a transcriptomic-based approach
2019-03-01 01:18FreericoBtistAllisonChurcherMnuelMnchoAlexnrLeitDeborhPower
Aquaculture and Fisheries 2019年1期
Freerico M.Btist ,Allison M.Churcher,Mnuel Mncho ,Alexnr Leit~o ,Deborh M.Power
a Centro de Ci^encias do Mar(CCMAR),Universidade do Algarve,Campus de Gambelas,8005-139 Faro,Portugal
b Instituto Portugu^es do Mar e da Atmosfera,Estaç~ao Experimental de Moluscicultura de Tavira,Portugal
c Department of Biosciences,College of Science,Swansea University,Swansea,SA2 8PP,Wales,United Kingdom
d IFAPA Centro El Toru~no,Junta de Andalucía,Camino Tiro Pichˊon s/n,11500 El Puerto de Santa María,Cˊadiz,Spain
e Environmental Science Center(ESC),Qatar University,P.O.Box:2713 Doha,Qatar
Keywords:Ruditapes decussatus Transcriptome Immune genes Toll-like receptors
A B S T R A C T The grooved carpet-shell clam Ruditapes decussatus is native to the Northern Atlantic and Mediterranean Sea and has a high commercial value.It is one of the main native bivalve species cultured in Europe.The main objective of the present study was to gain further insights into the immunological repertoire of R.decussatus through a transcriptomic approach.Pooled mantle samples of eight R.decussatus individuals were sequenced using Illumina platform.A total of 67 132 contigs with more than 800 bp were obtained.Manual annotation of these contigs revealed 146 immune-related genes.The gene families in which the highest number of immune-related genes was observed were:C1q domain-containing proteins(63),tumor necrosis factors(15)and toll-like receptors(TLRs,10).A total of 5 359 putative single nucleotide polymorphisms(SNPs)were identified in the 146 immune-related genes.The density of SNPs ranged between 0.04 and 7.92 SNPs/100 bp.The highest and the lowest SNP density were observed in genes of the C1q domain-containing protein family.Due to the importance of TLRs in innate immunity,we focused our attention on these membrane receptors.Ten TLRs were identified based on protein domain organization.Phylogenetic analysis revealed that R.decussatus TLRs were diverse and only 3 showed orthology with TLRs of known immune functions in other bivalve species.Moreover,our analysis suggests that lineage restricted-expansions of TLRs occurred in all mollusc taxa analysed including in venerids.
1.Introduction
Aquaculture is one of the fastest-growing food producing sectors in the world and has a high social and economic importance.According to the Food and Agriculture Organization(FAO,2014),the production of mollusc bivalves represented approximately 22%of world aquaculture production.The carpet shell clam Ruditapes decussatus is one of the main native bivalve species produced in Europe,with Portugal leading production in the world with a total production of 2 320 tonnes in 2012(FAO,2014).
Recently,there has been a considerable increase in knowledge about mollusc bivalve immunology motivated by the negative impact of infectious diseases on their production.High mortality rates have been observed in R.decussatus due to diseases caused by the protozoan parasite Perkinsus olseni and bacteria of the genus Vibrio(for review see Ruano,Batista,&Arcangeli,2015).As in other invertebrates,mollusc bivalves lack a specific immune response and associated immunological memory,and rely on the innate immune system.Several immune-related genes have been identified involved in the cellular and humoral innate response of bivalves,such as recognition of pathogen-associated molecular patterns,activation of signal pathways,haemocyte mediated reactions and production of immune effectors(Schmitt,Gueguen,Desmarais,Bach`ere,&de Lorgeril,2010).
The advent of next-generation sequencing(NGS)has greatly accelerated studies in the field of immunology of mollusc bivalves by providing insights into the genetic diversity and transcription of multiple genes simultaneously(Leite et al.,2013;Moreira et al.,2015;Zhang,Guo,Litman,Dishaw,&Zhang,2015).A better understanding of the immune system in R.decussatus will provide the basis for the development of strategies to mitigate the impact of infectious diseases affecting this species but can also be of relevance for other bivalves of commercial interest.The main objective of the present study was to map the immunological repertoire of R.decussatus using a transcriptomic approach.The importance of toll-like receptors in innate immunity led us to focus our attention on the identification and characterization of these immune-related membrane receptor genes.
2.Material and methods
2.1.RNA isolation,sequencing and de novo assembly
Total RNA was isolated from a piece of mantle edge from eight Ruditapes decussatus individuals(wild clams collected in Portugal)using the RNeasy Mini kit(QIAGEN).A Bioanalyzer 2100(Agilent technology)was used to assess the integrity of the RNA isolated.Total RNA from the 8 individuals was pooled and sequenced using an Illumina HiSeq 2000 platform at the Centre Nacionald'An`alisi Gen`omica(Barcelona,Spain)as reported in Benzekri et al.(2014).After adaptor removal and quality filtration(minimum Phred score of 20)the quality of the reads was confirmed with FastQC(Bioinformatics,2011)(see Fig.S1).After removing unpaired reads a total of 63 103 312 paired end reads(2×76 bp)were obtained.The GC content was 37%and the mean length and Phred quality were 76 bp and 36.3,respectively.Contigs were then obtained by de novo assembly using Trinity(release 2012-06-08)(Grabherr et al.,2011)with default parameters.
2.2.Annotation
The Annocript pipeline(Musacchia,Basu,Petrosino,Salvemini,&Sanges,2015)was used for annotation of R.decussatus contigs with more than 800 bp(a total of 67,132 contigs)using BLASTX against TrEMBL/UniRef and SwissProt(cut-off E-value≤10-5,threshold 18,wordsize 4),RPSBLAST against CDD profiles(cut-off E-value≤10-5),BLASTN against Rfam and rRNAs(cut-off E-value≤10-5).The Gene Ontology(GO)terms for cellular component,biological process and molecular function were obtained using Annocript.Conserved domains of immune-related genes were manually checked using SMART(Simple Modular Architecture Research Tool)(Letunic,Doerks,&Bork,2014)and transmembrane regions and signal peptide prediction were performed using Phobius(K¨all,Krogh,&Sonnhammer,2004)and SignalP 4.1(Petersen,Brunak,von Heijne,&Nielsen,2011),respectively.
2.3.Characterization of toll-like receptors
Contigs were identified as TLRs if they showed at least one leucine rich repeat(LRR)domain,a single transmembrane(TM)region and one C-terminal Toll interleukin-1 receptor(TIR)domain.Multiple alignments were carried out using Clustal Omega software(Li et al.,2015)with amino acid sequences from the TIR domain of R.decussatus TLRs identified in the present study,23 TLRs of Mytilus galloprovincialis(Toubiana et al.,2013),2 TLRs of Cyclina sinensis(Ren,Pan,Pan,&Bu,2016),12 TLRs of Lottia gigantea(Simakov et al.,2012),9 TLRs of Crassostrea gigas(Zhang et al.,2012,2013,2011;He et al.,2015)and 1 TLR of Chlamys farreri(Qiu,Song,Xu,Ni,&Yu,2007).The best amino acid substitution model was chosen using MEGA6 software(Tamura,Stecher,Peterson,Filipski,&Kumar,2013).Maximum Likelihood(ML)analysis were then conducted using MEGA6 and an LG substitution model and gamma distribution, five discrete rate categories and 1000 bootstrap replicates.Graphical representation of the phylogenetic tree was performed with iTOL(Letunic&Bork,2016).The RNA-seq reads of R.decussatus were aligned with the 146 immune-related genes using Burrows-Wheeler Aligner(BWA)-MEM(Li,2013).BWA-MEM was used with default settings,under which ambiguousalignments were randomly assigned to one of their possible positions of the immune-related genes.Samtools were then used to perform the SNP calling,which involved converting the SAM files into BAM files and sorting and indexing the BAM files.The samtools mpileup was used to collect summary information from the BAM files and compute the likelihood of the data and then convert it into BCF format.The bcftools were then used to filter the SNPs and generate a multi-sample variantcall format(VCF) file.Defaultparameters were used for both samtools mpileup and bcftools.
3.Results and discussion
3.1.Transcriptome characterization
A total of 173,927 contigs were assembled using Trinity with 67 132 longer than 800 bp.This subset of transcripts were selected for further characterization(mean sequence length of 2 349 bp).Successful Blast searches with at least one significant match (E<10-5)against UniProtKB/Swiss-Prot and UniProtKB/TrEMBL databases were achieved for 29 300 and 39 454 contigs,respectively,and at least one blast result was obtained for 40 839 out of the 67 132 contigs(longer than 800bp).The number of non-coding contigs was estimated to be 2 323.The functional categories distribution of the 40 839 annotated contigs using GO terms is shown in Fig.S2 of the supplementary data.We observed 3 913,1 510 and 757 GO functional categories belonging to biological process(BP),molecular function(MF)and cellular component(CC),respectively.These results suggest that the transcripts observed in the mantle of R.decussatus cover a wide range of biological processes.In the BP category,the “signal transduction”(GO:0007165)and the “transcription,DNA-dependent”(GO:0006351)terms were the mostabundant with 6.5 and 5.2%,respectively.In the MF category,the most abundant terms were “ATP binding” (GO:0005524), “zinc ion binding”(GO:0008270)and “metal ion binding”(GO:0046872)with 16.6,14.0 and 7.4%,respectively.The “integral to membrane”(GO:0016021)term was by far the most common one with 39.9%in the CC category.
3.2.Identification and characterization of immune-related genes
A total of 146 immune-related genes were identified in the transcriptome of the mantle of R.decussatus(for details see Table S1 of supplementary data).Thirty eight percent of the BLAST hits were obtained with members of the family Veneridae followed by 27%with members of the family Ostreidae(see Fig.S3 of supplementary data).The gene families in which the highest number of immune-related genes was observed were:C1q domain-containing proteins(63),tumor necrosis factors(15)and toll-like receptors(10)(Fig.1).The low number of immune-related genes may be explained by the fact that the individuals used in the present study were not challenged with pathogens and only mantle tissue was analysed.
C1q domain-containing proteins(C1qDCs)may function as pattern recognition receptors(PRRs)and participate in several immune responses.The sequencing and annotation of the genome of the Pacific oyster Crassostrea gigas revealed the presence of 321 C1qDCs,which is considerably higher than what has been reported in other protostomes and deuterostomes(Zhang et al.,2015).Zhang et al.(2015)reported that 164 C1qDCs were responsive to biotic stress in the Pacific oyster and hence highlighted their importance in the immune response.A more recent study by Lv et al.(2018)showed that 3 C1qDCs from C.gigas could bind lipopolysaccharide(LPS)and exhibited high binding activity towards Gram-negative bacteria.The high number of C1qDCs observed in the present study(64)are in line with the massive expansion of C1qDCs observed in other bivalves(e.g.Zhang et al.,2015).After C1qDCs,the highest number of expressed immune-related genes observed in the present study belonged to the tumor necrosis factor(TNF)family.TNFs are transmembrane proteins that among other functions,are involved in in flammation,apoptosis,cell proliferation and stimulation of the immune system(Goetz,Planas,&MacKenzie,2004).Initially,TNFs were presumed to be present only in deuterostomes,but more recently they were also identified in arthropods(Mekata et al.,2010)and molluscs(De Zoysa,Jung,&Lee,2009).Among the 15 TNFs identified in the present study,9 were predicted to have a typical TNF domain,namely a transmembrane region and a tumor necrosis factor domain(see Table S1).In C.gigas a total of 18 TNFs were identified in the whole genome(Zhang et al.,2015).One of them(CgTNF-1)was studied in more detail and is presumed to have a crucial role in the modulation of the immune response in apoptosis,phagocytosis and regulation of anti-bacterial activity(Sun et al.,2014).However,none of the TNFs identified in the present study were homologs of CgTNF-1,which suggest that R.decussatus may have more than 15 TNFs.Among the immune-related genes identified in the present study,it is also noteworthy the identi fication of lectins(4 C-type lectins and 5 galectins),big defensins(2)and lysozymes(3),which are presumed to be involved in the recognition and elimination of P.olseni(Soudant,Chu,&Volety,2013).
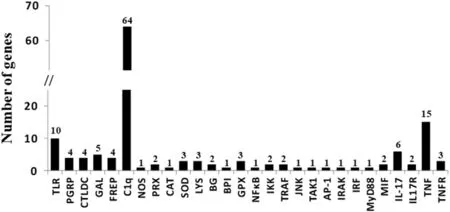
Fig.1.Immune-related genes expressed in the mantle of R.decussatus.Bactericidal permeability increasing protein(BPI),big defensins(BG),catalase(CAT),C-lectin domain containing proteins(CTLDC), fibrinogen domain containing proteins(FREP),galectins(GAL),globular head C1q domain containing proteins(C1q),glutathione peroxidases(GPX),inhibitor of nuclear factor kappa-B kinases(IKK),interferon regulatory factor(IRF),interleukins 17(IL-17),interleukin 17 receptors(IL17R),interleukin-1 receptor-associated kinase(IRAK),lysozymes(LYS),macrophage migration inhibitory factors(MIF),mitogenactivated protein kinase kinase kinase(TAK1),myeloid differentiation primary response gene 88(MyD88),nuclear factor kappa-B(NF-kB),nitric oxide synthase(NOS),peptidoglycan recognition proteins(PGRP),peroxiredoxines(PRX),stress-activated protein kinase(JNK),superoxide dismutase(SOD),TNF receptor-associated factors(TRAF),toll-like receptors(TLR),transcription factor AP-1 protein(AP-1),tumor necrosis factors(TNF);and tumor necrosis factor receptors(TNFR).
A total of 5 359 SNPs were identified in the 146 immune-related genes of R.decussatus.The mean density of SNPs in the 146 immunerelated genes was 2.17 SNPs/100 bp.The density of SNPs varied between 0.04 and 7.92 SNPs/100 bp(Fig.2).The level of DNA polymorphism observed(with one SNP every 46 bp)was similar to that previously reported by Sauvage,Bierne,Lap`egue,and Boudry(2007)in C.gigas and one of the highest described in metazoa.Among the 15 immune-related genes showing the highest SNP density,8 belonged to the C1q domain-containing protein family.Within each gene family,considerable variations in SNP density was observed.For example,the SNP density in TLRs varied between 0.07 and 3.58 SNPs/100 bp and the mean density was 3.28 SNPs/100 bp,which was higher than the mean density of all immune-related genes identified in the present study.
3.3.Toll-like receptors
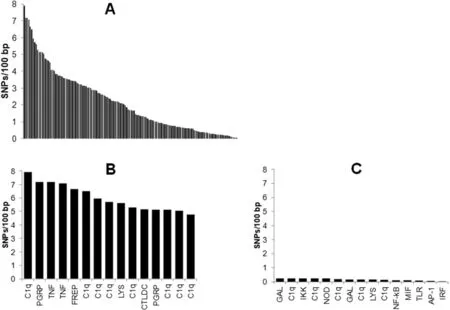
Fig.2.SNPs per 100 bp(in both coding and non-coding regions)detected in immune-related genes expressed in the mantle of R.decussatus.A,distribution of SNPs/100 bp in the 146 immune-related genes.B,the 15 genes with the highest number of SNP/100 bp.C,the 15 genes with the lowest number of SNP/100 bp.
Toll-like receptors(TLRs)are evolutionarily conserved pattern recognition receptors(PRRs)found in metazoans that have a key role in the innate immune system(Leulier&Lemaitre,2008).TLRs recognize pathogen-associated molecular patterns(PAMPs)present in pathogens such as fungi,bacteria and viruses.Gene expansion and functional divergence of TLRs was reported recently in bivalves(Zhang et al.,2015).The sequencing of the genome of C.gigas revealed the existence of 83 TLRs(Zhang et al.,2012,2015),although some of them were not predicted to have all the characteristic domains of TLRs(i.e.at least one LRR domain,one TM region and one TIR domain).The analysis of the transcriptome of other bivalve species also revealed a high number of TLRs namely,23 TLRs in the Mediterranean mussel M.galloprovincialis(Toubiana et al.,2013)and 32 TLRs in the Venus Clam Cyclina sinensis(Pan,Ren,Gao,&Gao,2015).In the present study,we identified 10 TLRs in the transcriptome of the mantle of R.decussatus(Fig.3).The number of LRRs in the 10 TLRs of R.decussatus,predicted using SMART,varied between 3 and 18(Fig.3).Four TLRs were predicted to have a signal peptide(RdTLR-2,-7-9 and-10).The absence of a predicted signal peptide in the other 6 TLRs may indicate that the nucleotide sequences are incomplete.Nevertheless,TLRs without signal peptides have been reported in other bivalve species with complete coding sequences namely in C.gigas(Zhang et al.,2015).Nine TLRs were single cysteine cluster TLRs(sscTLRs)and only one(RdTLR-2)was a multiple cysteine cluster TLR(mccTLR).
Our phylogenetic analysis suggests that multiple lineage restrictedexpansions may have occurred in bivalves such as in mytilids(clusters M1,M2 and M3)and venerids(cluster V1),and in the marine gastropod Lottia gigantea(cluster L1)(Fig.4).These results suggest that some TLR duplication events occurred early in mollusc evolution,but also more recently.Our phylogenetic analysis also shows that the TLRs of R.decussatus were diverse(Fig.4).The highest amino acid sequence similarity was observed between RdTLR-1 and RdTLR-6(51.4%)and the lowest between RdTLR-2 and RdTLR-8(7.3%).This contrasts with M.galloprovincialis in which higheramino acid sequence similaritieswere observed among some TLRs(Toubiana et al.,2013).For example,a high amino acid sequence similarity of 78,74 and 71%were observed for MgTLR-e/MgTLR-h, MgTLR-r/MgTLR-j and MgTLR-m/MgTLR-f,respectively.
Several TLRs of bivalves were shown to be differentially expressed following challenge with pathogens(bacteria,virus and fungi)or pathogen-associated molecular patterns(PAMPs)(He et al.,2015;Qiu et al.,2007;Ren et al.,2016;Toubiana et al.,2013;Zhang et al.2011,2013)(Fig.4).Zhang et al.(2015)observed that 19 out of 78 C.gigas TLRs were differentially expressed following challenge with Vibrio spp.,ostreid herpevirus 1 or LPS.Our phylogenetic analysis revealed that 3 TLRs of R.decussatus were orthologues with other bivalve counterpart with known immune function.RdTLR-2 clustered robustly with MgTLR-i with a boostrap value of 94%(Fig.4).Toubiana et al.(2013)found that MgTLR-i was significantly up regulated in haemocytes of M.galloprovincialis challenged with bacteria(V.anguillarum,V.splendidus or Micrococcus luteus)or fungus(Fusarium oxysporum).It is noteworthy that both RdTLR-2 and MgTLR-i are predicted to be mccTLRs.RdTLR-6 is an orthologue of CsTLR-4,which was upregulated in haemocytes of C.sinensi challenged with V.anguillarum or V.splendidus(Ren et al.,2016).Finally,our results also indicate that RdTLR-5 and CsTLR-13 are orthologues.Ren et al.(2016)observed that CsTLR-13 was up-regulated in haemocytes of C.sinensis following challenge with V.anguillarum,M.luteus or poly(I:C).Hence,it is likely that RdTLR-2,-5 and-6 have similar immune functions to their orthologues identified in M.galloprovincialis and C.sinensis.
4.Conclusions
A total 146 immune-related genes were identified in the transcriptome of the mantle of R.decussatus.The gene families in which the highest number of immune-related genes was observed were the:C1q domain-containing protein(64),tumor necrosis factor(15)and toll-like receptor(10)families.The density of SNPs in immune-related genes of R.decussatus ranged between 0.04 and 7.92 SNPs/100 bp.The highest and the lowest SNP densities were observed in genes of the C1q domaincontaining protein family.Based on amino acid sequence similarity and the predicted protein domain organization,we identified 10 TLRs in R.decussatus.The phylogenetic analysis of the TIR domain of R.decussatus TLRs revealed they were diverse and 3 TLRs(RdTLR-2,-5 and-6)were orthologues of TLRs from other bivalves with known immune functions.Moreover,our analysis suggests that lineage restrictedexpansions of TLRs occurred in most of the mollusc taxa analysed including the venerids.The present study increases our knowledge about the immunological repertoire of R.decussatus and sheds light on the evolution of mollusc TLRs and by analogy with other bivalve TLRs,their putative function.
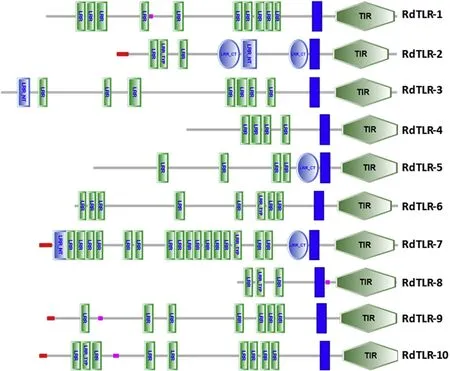
Fig.3.Toll-like receptors of Ruditapes decussatus.Red rectangle:signal peptide;Green rectangle:leucine-rich repeat(LRR)domain;Light blue rectangle:LRR N-terminal domain;Light blue ellipse:LRR C-terminal domain;Dark blue rectangle:transmembrane region;Purple rectangle:low complexity regions;Green hexagon:toll-interleukin 1 receptor(TIR)domain.(For interpretation of the references to colour in this figure legend,the reader is referred to the Web version of this article.)
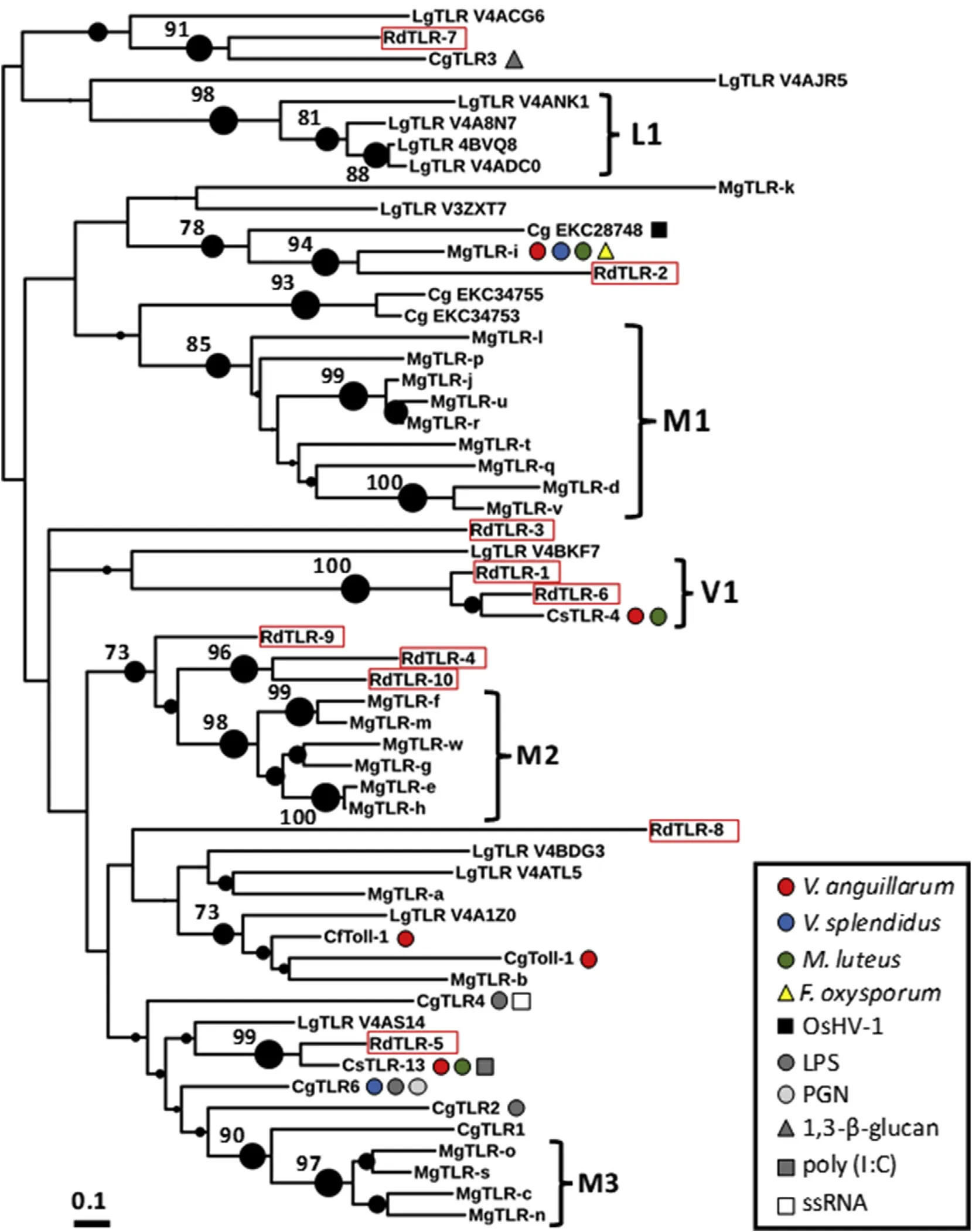
Fig.4.Phylogenetic tree of the TIR domain of toll-like receptors(TLRs)of Lottia gigantea(LgTLR),Ruditapes decussatus(RdTLR),Cyclina sinensis(CsTLR),Chlamys farreri(Cf),Mytilus galloprovincialis(MgTLR)and Crassostrea gigas(CgTLR)inferred through a maximum-likelihood analysis.TLRs of R.decussatus are indicated with a red rectangle.The size of the dark circles near nodes is proportional to the strength of bootstrap values.Only bootstrap values higher than 70%are shown near nodes.Symbols next to TLRs indicate they were differentially expressed following challenge with the gram-negative bacteria,Vibrio anguillarum(red circle)and V.splendidus(blue circle),the gram-positive bacterium,Micrococcus luteus(green circle),the fungus,Fusarium oxysporum(yellow triangle),the virus,OsHV-1(black square),lipopolysaccharide(LPS,dark gray circle),peptidoglycan(PGN,light gray circle),1,3-β-glucan(gray triangle),poly(I:C)(gray square)and ssRNA(white square).Differential expression data is based on the results of studies by Qiu et al.(2007),Zhang et al.(2011),Zhang et al.(2013),Toubiana et al.(2013),He et al.(2015)and Ren et al.(2016).(For interpretation of the references to colour in this figure legend,the reader is referred to the Web version of this article.)
Acknowledgements
This study was financially supported by the projects:AQUAGENET(SOE2/P1/E287)funded by the European Regional Development Fund within the program INTERREG IVB SUDOE;COMPETE/FEDER(FCOMP-01-0124-FEDER-010607);PTDC/MAR/103550/2008 and CCMAR/Multi/04326/2013 funded by the Foundation for Science and Technology(FCT).F.M.B.and A.M.C were supported by postdoctoral fellowship grants from FCT(SFRH/BPD/108591/2015 and SFRH/BPD/85408/2012).We would like to thank two anonymous referees for their constructive comments.
Appendix A.Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.aaf.2018.03.003.
 Aquaculture and Fisheries2019年1期
Aquaculture and Fisheries2019年1期
- Aquaculture and Fisheries的其它文章
- Genome-wide association study identifies loci for body shape in the large yellow croaker(Larimichthys crocea)
- Sex-biased gene discovery from the gonadal transcriptomes of the large yellow croaker(Larimichthys crocea)
- SuperSAGE digital expression analysis of differential growth rate in a European sea bass population
- Molecular aspects of eye development and regeneration in the Australian redclaw cray fish,Cherax quadricarinatus