
偏二甲肼与二氧化氮气相反应的数值模拟研究
2019-02-19 07:17崔村燕周宵灯辛腾达韩向阳
导弹与航天运载技术 2019年1期
詹 翔,崔村燕,周宵灯,辛腾达,韩向阳
(航天工程大学,北京,101416)
0 引 言
目前,中国在航天领域使用的大型火箭所采用的液体推进剂主要是肼类燃料(偏二甲肼UDMH)和硝基氧化剂(四氧化二氮 NTO),具有很强的毒性、腐蚀性、吸湿性和易燃、易爆、易挥发等特点[1]。由于推进剂在贮存过程中采取了一系列防护措施,发生大规模泄漏的可能性较小,容易出现的泄漏问题主要是渗漏或滴漏。渗漏或滴漏过程中,偏二甲肼主要以蒸汽形式扩散,四氧化二氮则分解为二氧化氮扩散。当两者相遇时发生剧烈燃烧、爆炸,需要温度和浓度条件如下[2]:
a)25 ℃以下,9%气相偏二甲肼与 100%气相二氧化氮;
b)50 ℃以下,61%气相偏二甲肼蒸气与 35%气相二氧化氮接触;
c)70 ℃以上,两种推进剂蒸汽接触。
由此可见,偏二甲肼与二氧化氮同时泄漏后发生反应不会立即爆炸,但两者发生缓慢化学反应所释放的热量有可能触发爆炸。本文通过数值模拟的方式研究常规推进剂间的气相反应规律,为相关事故处理措施提供依据。
1 反应机理
Ernesto等[3]通过试验研究给出了偏二甲肼与二氧化氮在无光照、室温(15 ℃)、气相条件下的反应过程。所有反应物和生成物均被傅里叶变换红外光谱仪进行检测。试验中各组分浓度随时间的变化如表1所示。

表1 试验中相关物质在各时刻的浓度Tab.1 Concentration of the Related Substances at All Times in the Test

续表1
Ernesto等人根据试验产物给出如下反应路径:


由试验产物反应可以看出,在室温条件下偏二甲肼与四氧化二氮分解产生的二氧化氮气相反应较为缓和,二氧化氮夺取偏二甲肼上与氮原子相连的氢原子,生成中间产物(B)以及亚硝酸(HONO);在无光照和室温条件下,中间产物(B)在二氧化氮的作用下进一步脱去氮上的氢原子生成(H),此时中间体(H)上的氮原子相连接的氢全部被二氧化氮夺走,氮原子上存在两个未配对的电子。当两个(H)相遇时,有未配对电子的氮原子就会配对形成氮氮双键,形成最终产物(I)1,1,4,4-四甲基-2-四氮烯。结合反应物消耗速率和生成物合成速率,给出如下反应方程式:

其整体反应速率系数由肼的衰变率确定为2.3×10-17cm3/(mol·s)。该试验在无光照、室温 15 ℃条件下进行,是绝大多数液态推进剂贮存的环境条件。因此该反应机理可以代表偏二甲肼与四氧化二氮推进剂泄漏时的气相反应过程。
基于FLUENT软件模拟化学反应时,放热量是根据生成物与反应物的生成焓之差计算得到的。1,1,4,4-四甲基-2-四氮烯(TMTZ)是一种含氮化合物,分子结构如图 2所示,常见于偏二甲肼和二氧化氮的反应产物中[4~6]。为获得 TMTZ分子的生成焓,采用量子化学的方法对TMTZ分子的生成焓进行计算。

图2 TMTZ分子结构示意Fig.2 TMTZ Molecular Structure Diagram
2 TMTZ分子生成焓计算
2.1 计算方法
生成焓对于高能物质而言是十分重要的一项,是评价物质安全性和能量水平的标准之一[7]。目前量子化学方法能十分精确地计算高能化合物的能量,并且采用理论计算可以避免重复大规模试验带来的经济负担以及试验过程中可能发生的毒害和爆炸危险。通过量子化学的方法计算得到 TMTZ的生成焓既经济又安全。骆艳娇等[8]对几种不同多氮含能材料的生成焓进行了计算,同时对比了几种不同的密度泛函方法,研究过程表明:对于多氮含能材料,杂化密度泛函方法(DFT-B3LYP)计算出的生成焓可靠度高、占用资源少,计算速度快。本文采用的密度泛函理论方法计算生成焓,存在计算结果不直接给出生成焓,只能得到分子的一些热力学参数,要借助辅助方法才能求得目标化合物的生成焓。常用的辅助方法包括原子化反应法[9~11]、高斯理论法等。
高斯理论法(Gaussian theory)是目前使用较为广泛的理论计算化合物能量的方法之一。1989年John等人发表 Gaussian-1(简称 G1)方法以来,已经有 G2、G3和G4等4代方法。在缺少试验值的情况下,其计算结果有时也被当作标准数值使用[12]。若分子体系中的非氢原子超过7个,高斯理论方法计算耗时将大大增加。而TMTZ分子中的非氢原子为7个,计算量将成指数级增加,因此不适宜使用高斯理论法。
基于上述分析,本文采用原子化反应法对热力学参数进行处理以获得TMTZ分子的生成焓。
2.2 生成焓计算
选用含有电子相关效应的B3LYP杂化密度泛函关联 6-31G(d)基组对 TMTZ分子结构进行几何优化计算,获得了能量低、稳定的构型,结果如图3所示。
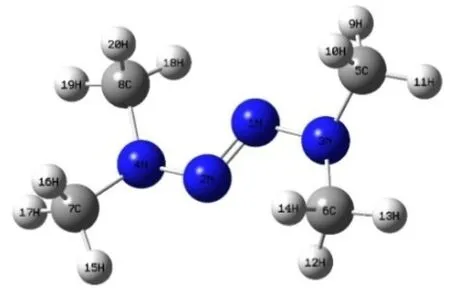
图3 TMTZ最稳定分子构型示意Fig.3 Stable Molecular Configuration of TMTZ
在稳定构型的基础上进行频率计算,获得焓的热校正量Hcorr和零点矫正能εZPE;分别在B3LYP杂化密度泛函关联6-31G(d)基组下计算TMTZ分子中H、C、N的单个原子总能量,并计算分子的总能量。将计算结果代入原子化反应公式[13,14],有:
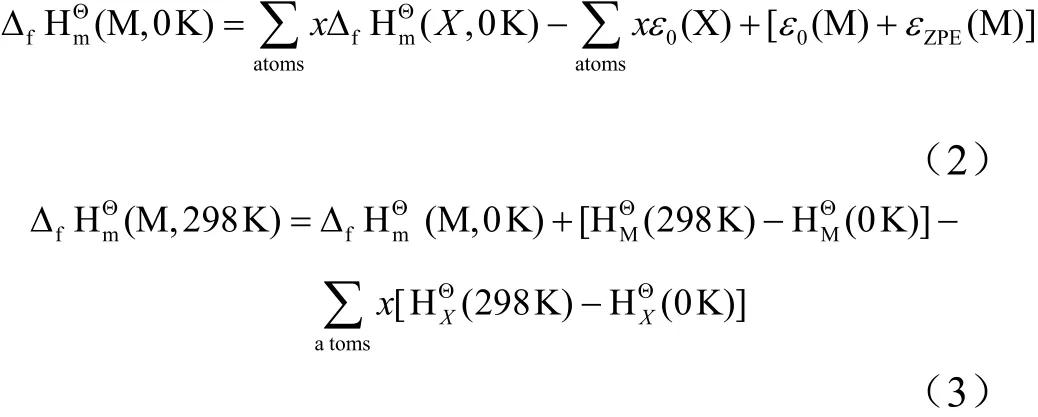
式中 M为分子;X为M分子的组成原子;Δf(X,0K)为各组成原子生成焓;ε0(X)为各组成原子总能量之和;[ε0( M) + εZPE(M)]为M分子的单点能加上其零点矫正能;(298K )- Δ HΘ(0K)为M分子的生成焓温度修正值且 f m
等于 Hcorr-εZPE、[( 298K)-(0K)]各原子的焓值温度修正值之和。
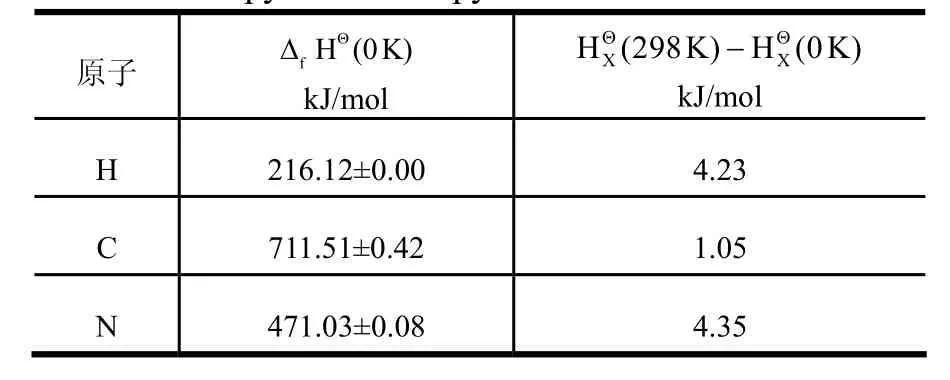
表2 几种气态原子在0K时的生成焓值和生成焓矫正值[15]Tab.2 Enthalpy and Enthalpy of Formation of Atoms at 0K
基于量子化学方法计算出H、C、N原子的单点能ε0( X)、分子的焓值差( 298K)-(0K)、单点能ε0(X)以及零点能εZPE(M)之和,结果如表3所示。
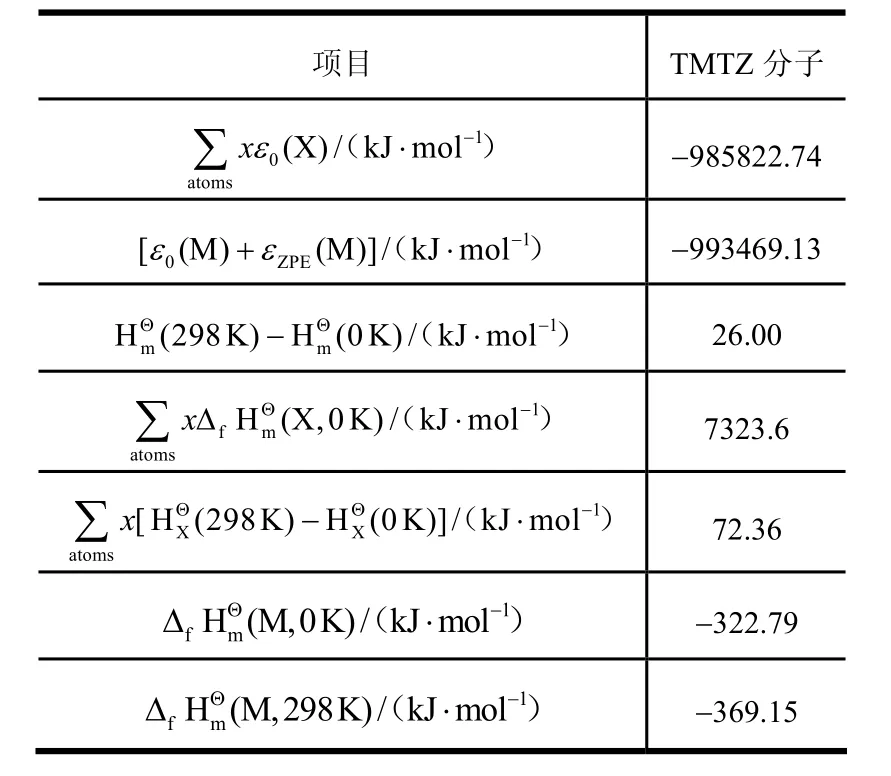
表3 TMTZ分子计算结果Tab.3 Calculation Results of TMTZ
密度泛函理论是目前应用较为广泛的量子化学方法,采用B3LYP杂化密度泛函计算多氮含能物质生成焓的误差平均为[8]18.84 kJ/mol。对于TMTZ分子来说,理论误差约占计算值的5%,占比较小。该计算结果具有一定的可靠度,可用来计算化学反应的放热量,分析反应过程中温度的变化趋势。
3 数值模拟过程
3.1 计算域和基本设置
气相反应试验在1.2 m×2.4 m×2.4 m的箱式密闭容器中进行[3]。计算域根据试验采用2.4 m×2.4 m的2D模型,四周采用壁面条件。反应条件为15 ℃、一个标准大气压。假设反应物通入到容器中后立即完全混合,在数值模拟初始化时直接根据表1设置反应物的预混浓度。反应的背景填充物为空气。
3.2 数值模拟与试验结果对比
设置监测面记录反应物、生成物在计算域中的平均浓度变化,计算结果如图4所示。从图4中可以看出,反应速率根据反应物浓度经历了一个由快到慢的过程,开始时,由于反应物浓度较大,反应速率较快,当反应进行到5 min左右时,反应物的浓度减少导致反应速率下降,生成物浓度几乎不再变化。根据计算结果,生成物、反应物的增减关系严格遵循式(1)给出的化学计量数。
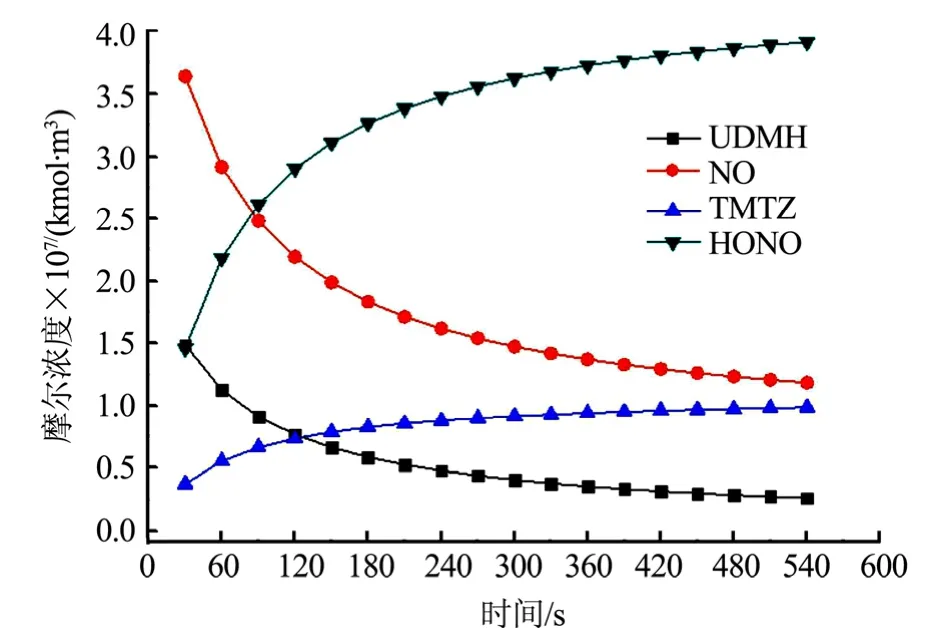
图4 各物质摩尔浓度随时间变化曲线Fig.4 Molar Concentration of Each Component with Time
各组分试验值和数值模拟结果对比如图5所示。

续图5
从图5中可以看出,数值模拟结果与试验值在变化趋势上基本保存一致。为了定量地研究数值模拟结果与试验结果的差异,采用Hanna等人提出的误差估计方法评价的模拟效果[16,17]。该方法的误差分析包括F(平均分数偏差)和M(几何平均偏差)、V(几何平均方差)、N(标准化均方差)和FAC2(模拟值与观测值之比),分别表示为

式中 Xo为试验观测值;Xp为模拟值。
该误差统计分析法可以较为全面地比较模拟结果与试验值的差异[18]。通过以上分析方法对数值模拟结果进行统计,结果如表4所示。

表4 各组分数值模拟结算误差分析Tab.4 Numerical Simulation’s Deviation of Each Component
对于可靠性较高的数值模拟结果,其误差分析得到的各参数应满足[19]:-0.67<F<0.67,N<4,FAC2>0.5。由表4可知,数值模拟结果的误差均在合理的区间内,数值仿真较好地复现了试验过程。从图5对比中分析,对于反应物来说,数值模拟计算的结果总是略高于试验值,说明在实际反应过程中,还有少量的UDMH和NO2转换成了其它物质。由文献[20]的分析可知,生成物可能是二甲基亚硝铵(CH3CH3N-N=O)、二甲胺(CH3CH3N-H)、甲醇(CH3OH)、二甲基甲酰胺(CH3CH3N-CH=O)和二甲基肼硝酸盐(CH3CH3N-NH3NO3)等甲基衍生物。从偏二甲肼的浓度误差来看,该转化量比较少,在误差可接受的范围内。结合生成物浓度情况,HONO的浓度值总是小于试验值,说明生成物HONO还有其它合成来源,可能是NO2与空气中的组分发生了一系列反应生成的额外的亚硝酸HONO。
设置监测点对反应区域温度进行检测,取整个区域的平均温度,结果以288 K为零点,如图6所示。

图6 温度随时间变化曲线Fig.6 Temperature Along with the Time Variation
由图 6可知,在试验给出的反应物浓度下,反应放热使气体升温的效果不明显。数值模拟结果显示:温度增长趋势与反应速率增长趋势一致,反应释放的热量使整个区域温度上升约0.072 K。但是在实际泄漏的情况下,由于偏二甲肼与二氧化氮均比空气重,两推进剂蒸汽受到重力的影响在地势较低的地方聚集。当积累的浓度较高时,反应释放的大量热会使推进剂间的反应类型由冷反应阶段(该反应阶段默认反应温度在室温 15 ℃左右)转变为热分解阶段[21]。在热分解阶段,一些大分子物质可能受到高温的影响发生热分解反应放出大量热,进一步加速热分解反应,使推进剂蒸汽发生剧烈燃烧、爆炸。
4 结 论
a)采用DFT-B3LYP方法计算出TMTZ分子的热力学数据,经原子化反应法处理后得到标准生成焓,为-369.15 kJ/mol;
b)数值模拟结果与试验值基本一致,反应速率增长趋势相同,数值仿真能较好地反映生成物浓度变化;
c)计算结果与实验结果存在偏差的原因是:少量偏二甲肼与二氧化氮反应生成了其它甲基衍生物,同时二氧化氮与空气中的组分反应生成了额外的亚硝酸;
d)反应释放的热量使整个区域温度上升了约0.072 K。
猜你喜欢
军民两用技术与产品(2022年7期)2022-08-06
装备环境工程(2022年2期)2022-03-15
家庭医学(2021年6期)2021-07-19
兵器装备工程学报(2020年11期)2020-12-16
固体火箭技术(2020年4期)2020-09-05
火炸药学报(2019年2期)2019-05-05
中学化学(2017年5期)2017-07-07
科技与创新(2017年9期)2017-06-07
绿色科技(2017年6期)2017-04-20
中学化学(2016年4期)2016-05-30
