
大DNA体内组装技术进展
2019-02-15 08:31王培霞
生物加工过程 2019年1期
王培霞,马 渊,吴 毅
(1. 天津大学 化工学院 系统生物工程教育部重点实验室,天津 300072;2. 天津大学 天津化学化工协同创新中心 合成生物学研究平台,天津 300072)
随着测序技术的发展,人类对遗传信息的读取认知呈爆炸式增长,与此同时,编写DNA的尺度也不断延伸,逐渐由单个基因、某一代谢通路向完整基因组拓展。研究对象复杂性的提升对大尺度DNA的合成与组装提出了更高的需求。由于大尺度DNA在体外操作时极易断裂,因此,组装100 kb以上的DNA分子一般选择在细胞内进行[1]。基于酿酒酵母、大肠杆菌和枯草芽孢杆菌这些模式生物自身的高效重组机制,研究者开发了一系列的大尺度DNA的体内组装技术,已经用于合成与组装外源代谢途径、病毒基因组、细菌基因组、酵母基因组和高等生物某些基因或染色体区域。本文中,笔者综述基于大肠杆菌、酿酒酵母和枯草芽孢杆菌的体内大DNA组装技术的研究进展,为研究者提供参考和借鉴。
1 基于大肠杆菌重组机制的组装技术
大肠杆菌存在天然的重组系统——RecA重组系统[2]。RecA重组系统由RecA和RecBCD组成,RecA蛋白促进2个DNA分子之间的同源联合和置换,RecBCD是RecB、RecC和RecD组成的复合体,依赖于ATP发挥核酸外切酶和解旋酶作用。RecBCD与双链DNA分子末端结合,使DNA双链解链形成单链,然后由RecA蛋白催化进行同源重组。在实际应用中,RecA重组系统效率较低,因此其应用有限。1998年,Zhang等[3]报道了一种更高效的大肠杆菌体内重组系统——λRed/ET重组系统,其中,λRed重组系统由λ噬菌体中的3个基因exo(Redα)、bet(Redβ)和gam(Redγ)组成[4],Redα具有5′→3′方向的核酸外切酶活性,从5′端切割双链DNA分子,产生3′端突出末端。然后单链DNA结合蛋白Redβ和RecT结合到3′突出末端,一方面保护单链DNA末端不被细胞内的单链核酸酶降解,另一方面能介导外源单链DNA退火,导致DNA分子同源区域的复制和交换,从而发生重组。Redγ蛋白结合大肠杆菌自身的RecBCD复合蛋白,抑制RecBCD的核酸外切酶活性,防止宿主对外源线性DNA的降解[5]。λRed/ET重组系统的同源序列只要达到50 bp,就能实现片段与片段、片段与载体以及片段与基因组之间的同源重组。此外,将λRed/ET重组系统和Cre/loxP、FLP/FRT等位点特异性重组系统相结合,可以实现DNA元件的组装、克隆、置换、敲除和突变[6-7]。
2005年,Wenzel等[8]利用RecET重组系统在大肠杆菌中重建了来自枳实(Stigmatellaaurantiaca)的黏液色素S生物合成的基因簇(长度为43 kb),其中包含了在异源宿主假单胞菌中表达所需的基因元件。2007年,Smailus等[9]利用λRed重组系统开发了一种载体系统,利用细菌F-质粒载体在大肠杆菌体内迭代组装大DNA分子,构建2种F-质粒载体实现抗生素转换。利用λRed重组系统介导的迭代组装,成功重建了流感嗜血杆菌(Haemophilusinfluenzae)基因组的2个非连续区域,总长度190 kb,占流感嗜血杆菌基因组的10.4%。该方法原则上可用于构建完整的流感嗜血杆菌基因组。2012年,Stewart研究团队的Fu等[10]利用RecET重组系统介导的高效同源重组机制,将发光肠杆菌(Photorhabdusluminescens)的所有巨型合成酶基因(每个长度为10~52 kb)直接克隆到大肠杆菌的载体上,并在异源宿主中表达了其中的2个基因。由此可见,λRed/ET重组系统操作简单,是在大肠杆菌中组装长片段DNA的有力工具,由于宿主易培养、易扩增、易分子操作,使得λRed/ET大肠杆菌重组系统具有组装周期短、方便转移到其他表达宿主等优点。
2 基于酿酒酵母同源重组机制的组装技术
酿酒酵母具有从体外吸收并在体内组装DNA片段的能力,并且其同源重组效率非常高,常常用于体内多片段DNA的组装[11-13]。编码整个异源的代谢通路通常需要组装多个基因片段,利用酵母体内同源重组可以一步组装整个代谢通路[14],组装效率高达70%~100%。2015年,Guo等[15]开发了YeastFab组装方法,利用该方法可以一步将标准化的生物元件组装为转录单元,并且利用多种启动子元件的组合组装来优化代谢通路。染色体和基因组的合成也广泛使用了酵母组装技术。2006年,美国科学院院士Jef.Boeke发起人工合成酵母基因组计划(Sc2.0),旨在化学合成世界上首个真核生物基因组。酿酒酵母16条染色体的合成正是依赖于自身高效的同源重组机制。酵母长染色体的合成是一个多层次逐级组装的过程,首先由化学合成的单链DNA体外组装为750 bp的双链DNA砖块(building blocks),并且在体外进一步组装成2~3 kb的小模块(minichunks),或者继续组装成8~10 kb的大模块(megachunks),然后一次将6~12个模块导入酵母体内,利用同源重组完成大片段DNA组装以及对应野生型染色体序列的替换。通过多轮的顺序组装与替换,最终实现完整合成型染色体的组装,并且替换掉原有的野生型染色体[16]。
3 基于枯草芽孢杆菌重组机制的组装技术
枯草芽孢杆菌具有吸收外源DNA的能力,外源质粒在枯草芽孢杆菌内复制时会出现不稳定的单链DNA形式,导致质粒的丢失,一般通过RecA介导的同源重组将外源DNA整合到宿主染色体上以实现外源DNA的稳定[22]。1995年,Itaya[23]采用枯草芽孢杆菌基因组(BGM)作为DNA克隆的载体,组装了长达48.5 kb的大肠杆菌原噬菌体的λDNA。2000年,为了测试枯草芽孢杆菌载体系统构建大尺度DNA的能力,Itaya等[24]和Iwata等[25]将长度约120 kb的小鼠基因组DNA克隆到枯草芽孢杆菌的基因座中,并且可以维持小鼠DNA的稳定性。基于BGM载体系统,通过两段小DNA序列(LPS)的定位和定向结合发展了长片段DNA克隆方法——尺度延伸法(inchworm elongation method)。研究者利用这种组装方法组装了光合细菌蓝藻PCC6803的基因组,成功把3.5 Mb的蓝藻PCC6803基因组整合到4.2 Mb的枯草芽孢杆菌BGM载体上形成一个7.7 Mb的杂合基因组[26]。由于尺度延伸法需要高纯度的长片段(超过100 kb)的DNA模板,一定程度上限制了该方法的应用。除此之外,BGM载体系统还存在载体同源序列之间非理性重组的问题。
为了提高尺度延伸法的组装效率以及可操作性,Itaya等[27]开发了多米诺骨牌法(domino method)组装技术。多米诺骨牌法通过两种抗生素标记基因的交替使用,在BGM载体中进行多轮多米诺片段延伸来组装超大DNA片段。Itaya等[27]利用多米诺骨牌法成功将16.3 kb的小鼠线粒体基因组和134.5 kb的水稻叶绿体基因组整合到BGM载体上。为减少组装大片段所需步骤,需要增加多米诺骨牌DNA的长度,可以利用大肠杆菌来源的细菌人工染色体(BAC)提供长度约为100 kb的DNA片段用于多米诺骨牌法[28]。以枯草芽孢杆菌为宿主开发的组装技术仅能稳定维持GC含量在44%以下的外源大DNA片段(长度大于200 kb)[28]。此外,为了避免非理性重组问题,Ogawa等[29]开发了RecA诱导型BGM载体(iREX),使RecA的表达受到培养基中木糖的控制,在缺乏木糖的条件下,RecA在iREX系统中不表达,相反,存在木糖的条件下则诱导RecA表达,使iREX能够发挥与BGM载体相同的组装能力,提高了BGM载体系统的严谨性。
4 体内组装技术用于合成基因组学
基因组学研究由“读”到“写”的转变造就了合成基因组学领域的萌发和快速发展。合成基因组学通过对基因组进行理性设计与化学合成,实现改造生命形式和重塑生命功能。纵观合成基因组学的发展历程,合成基因组学的研究对象实现了由病毒到原核生物再到真核生物的发展,设计的复杂度也由简单的复制序列发展为多元化甚至颠覆性的基因组重塑。
4.1 病毒基因组合成
2002年,Cello等[30]在没有天然模板的情况下化学合成了脊髓灰质炎病毒cDNA。脊髓灰质炎病毒基因组是合成基因组研究的开端[31]。从长度约为69 nt的寡核苷酸出发,利用其末端具有重叠的互补序列来合成400~600 bp的短片段,然后将片段连接到质粒载体中,再进一步酶切连接成长片段。最后通过酶切连接3个长片段,得到全长的脊髓灰质炎病毒cDNA。通过此方法化学合成的病毒cDNA成功转录成病毒RNA,产生具有感染性的病毒。尽管脊髓灰质炎病毒cDNA大小仅为7 740 bp,科学家却花费了数个月的时间来化学合成其基因组。J.Craig Venter实验室改进了之前的方法,开发了自动化的数字生物转换器,明显缩短了从合成寡核苷酸到组装5~6 kb片段所需的时间,采用该方法,Venter实验室的Smith等[32]仅用两周时间组装了长度5 386 bp的完整噬菌体φX174基因组[32]。
上述病毒基因组合成均是对野生型基因组序列的严格合成,为了实现不同的研究目标,可能需要对基因组进行人工设计。2005年,Chan等[33]为了实现基因组的物理分离并实现对某些遗传元件的单独操纵,重新设计了T7噬菌体的基因组,并且将基因组的左边11 515 bp的序列替换为12 179 bp的设计序列,由此产生的半合成型噬菌体能够存活且和野生型噬菌体无异,该研究表明了重新设计和构建天然生物体基因组的可行性。
4.2 细菌全基因组合成
2008年,Fraser等[34]完成了生殖道支原体(Mycoplasmagenitalium)基因组的人工合成,命名为JCVI-1.0。这个人工合成的基因组全长582 970 bp,含有野生型生殖道支原体G37的所有基因(除MG408外)。生殖道支原体是能在自然条件下生长的最小的生物体,其基因组大小是φX174的100倍。合成JCVI-1.0基因组以5~7 kb的基因片段为原材料,体外重组法逐步组装长度更大的中间体,将中间体组装在大肠杆菌的BAC上。最后,通过酿酒酵母组装成完整的合成型基因组[35]。此方法成功构建了正确的全基因组序列,但是组装过程更多地依赖于体外酶促反应,操作繁琐,而且从酵母中提取组装的大片段DNA效率较低。2010年,Lartique等[36]利用酵母同源重组完成了长度为1.08 Mb的蕈状支原体(Mycoplasmamycoides)JCVI-syn1.0基因组的设计与合成,并将其成功移植到受体细胞山羊支原体(Mycoplasmacapricolum)中,产生新的支原体细胞,细胞中含有的DNA全部为合成型DNA序列,新细胞具有预期的表型特性,并且能够连续自我复制[37]。2016年,在JCVI-syn1.0的基础上,Hutchison等[38]通过不断再设计、合成和测试,产生了更加精简的合成型基因组JCVI-syn3.0(长度531 kb,包含473个基因),这对于研究生命的核心功能和探索全基因组设计合成有重大意义。JCVI-syn3.0的组装过程与JCVI-syn1.0基本相同,都是借助了酿酒酵母高效的同源重组系统,但是在酵母中提取大DNA片段用于下一步组装时,两者采用了不同的技术。JCVI-syn3.0利用了滚环扩增(rolling circle amplification)的方法取代了在JCVI-syn1.0中低效的脉冲场凝胶片段回收方法。
已经组装成功的蕈状支原体基因组具有不采用通用遗传编码的特点,其UGA编码色氨酸而非终止密码子,该特性避免了对组装宿主酵母产生细胞毒性。在另一项工作中,Karas等[39]合成1.5 Mb的无胆甾原体(Acholeplasmalaylawii)PG-8A的基因组,该基因组使用通用遗传密码,发现有一个基因的克隆对酵母具有毒性,导致无法组装出全长的无胆甾原体基因组。通过灭活该基因,可以维持无胆甾原体基因组在酵母细胞中的稳定遗传[39]。此外,海洋蓝细菌原绿球藻(Prochlorococcusmarinus)MED4基因组也在酵母中实现组装,该基因组长度1.66 Mb,GC含量和酿酒酵母相似,且基因组上均匀分布了酵母复制起始位点序列(ARS),研究发现该基因组能够稳定遗传[40]。Heuer等[41]克隆GC含量为66%的铜绿假单胞菌(Pseudomonasaeruginosa)染色体,成功克隆的DNA片段大小均小于120 kb。构建GC含量为69%的黄色黏球菌(Myxococcusxanthus)线性酵母人工染色体(YAC)文库,成功插入的DNA片段大小为40~180 kb。组装基因组GC含量为55%的细长聚球蓝细菌(Synechococcuselongatus)PCC 7942,发现在酵母中不能稳定维持超过约200 kb的DNA片段,通过添加酵母复制起点序列,使得组装长度达到454 kb以上。由此可见,酵母可以组装并稳定维持GC含量在32%~38%之间的细菌基因组序列[37,39-40],但是高GC含量可能会限制酵母克隆DNA的长度,利用酵母组装高GC含量的长片段DNA序列时,应在合成序列中额外添加酵母复制起始位点作为维持外源DNA稳定的元件[42]。
4.3 真核全基因组合成
在人工合成酵母基因组计划中,Gibson[13]提出了三项设计原则:1)包含合成型染色体的细胞应与野生型细胞有相似的表型和适应性;2)通过删除一些不稳定的序列和元件,增加合成型基因组的稳定性;3)通过引入合成生物学元件增加合成型基因组的遗传操作灵活性。在此设计原则基础上,合成型酵母的基因组与野生型基因组相比缩减了约8%,有约1.1 Mb的序列被删除、插入或者修改。2011年,研究者率先合成了一部分的真核生物染色体——酿酒酵母Ⅸ号染色体右臂(synIXR)和Ⅵ号染色体左臂(synVIL)[13]。2014年,Annaluru等[43]报道了首条完整的合成型真核生物染色体——合成型三号染色体(synIII),从野生型长度为316 617 bp酿酒酵母三号染色体出发,最终合成1条有功能的长度缩短为272 871 bp的synIII。
2017年,Sc2.0团队完成了酿酒酵母synII、synV、synVI、synX和synXII共5条合成型染色体的从头设计与合成[44-48],宣布合成型酵母基因组1/3的工作完成。Shen等[44]报道了完成长度为770 035 bp的酿酒酵母二号染色体的合成,并且使用多组学的手段(表型组、转录组和蛋白组)综合表征了synII。Xie等[45]报道了精确匹配设计序列的长度为536 024 bp的酿酒酵母五号染色体的合成,并且构建了一个环形synV的衍生形式,研究人员使用共转化和CRISPR/Cas9修复了22处碱基突变,构建的synV菌株在各种检测条件下与野生型菌株相比都没有显著差异。Mitchell等[46]报道了设计、组装和表征长度为242 745 bp合成型六号酵母染色体synVI,研究人员还首次将3条合成型的染色体(synIII、synVI和synIXR)组合在一个酵母细胞内,虽然包含3条合成型染色体的酵母有轻微的长势问题,但是极大地推动了全合成酵母基因组的进程。Wu等[47]报道了化学合成了设计长度为707 459 bp的酿酒酵母十号染色体(synX),并且开发了一种命名为混菌PCR标签定位(pooled PCR Tag mapping[PoPM])的高通量定位策略用于高效识别合成型染色体组装过程中出现的生长缺陷靶点(bug),排除缺陷靶点之后的合成型酵母十号染色体菌株在测试的各种培养条件包括高灵敏度的竞争生长实验中都展示了与野生型很相似的生长状态。Zhang等[48]报道了设计合成长度为976 067 bp的合成型十二号染色体synXII,使用合成型DNA片段迭代替换和减数分裂同源重组2种策略结合的方式,组装了全长的synXII,研究人员还将酿酒酵母十二号染色体上的核糖体基因簇(rDNA)区域删除并且在染色体其他3处位置引入贝酵母(Saccharomycesbayanus)的rDNA序列。Sc2.0项目的阶段成果奠定了对更大尺度、更复杂结构的基因组进行设计与编写的基础。表1总结了大DNA组装的应用实例。
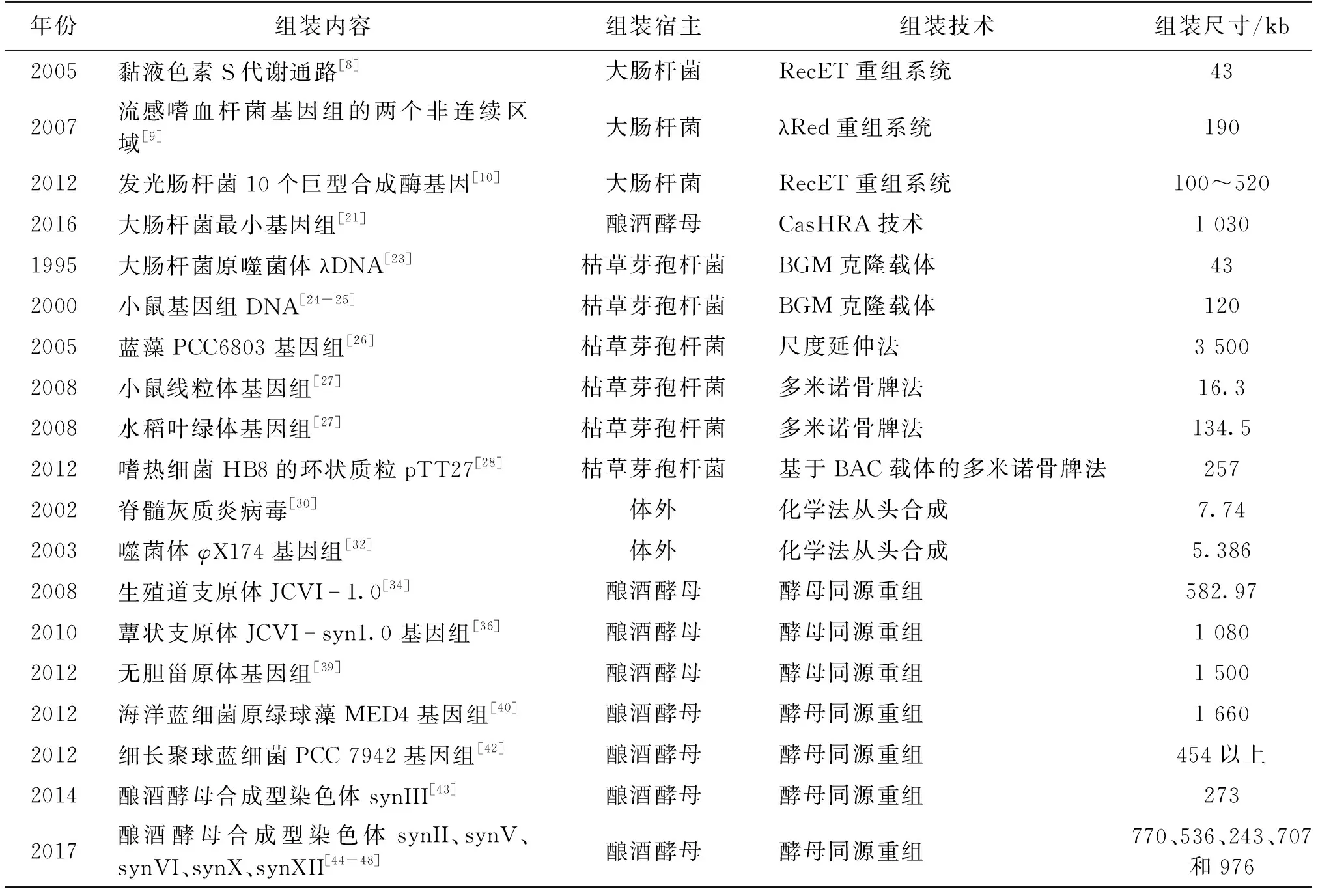
表1 大DNA组装汇总
此外,Karas等[49]还在酵母中进行了其他真核生物染色体的组装工作。以TAR(transformation associated recombination)克隆技术得到的约100 kb DNA片段出发,三角褐指藻(Phaeodactylumtricornutum)的25和26号染色体(长度分别为497和441 kb)在酵母体内完成组装,研究发现,添加酵母复制起点序列能够改善酵母中大DNA的稳定维持,且纯化待组装的DNA片段可以提高组装效率。此外,超过1 Mb的人类淋巴母细胞DNA以及长达2.3 Mb的大尺度DNA已经在YAC上成功组装,并且实现哺乳动物细胞的转染[50-51]。
尽管DNA的测序以及编辑技术在快速发展,但是在细胞中构建和表达大尺度DNA的过程却受到了不少限制,这阻碍了对生物系统的全面理解和改造能力。2016年6月,Boeke等[52]宣布合成基因组学的旗舰项目——基因组编写计划(Genome Project-Write(GP-Write))正式启动。基因组编写计划是一个开放的、国际合作项目,由多个学科领域的科学家共同参与,包括生物学、化学、计算生物学、工程、社会科学和生物伦理学。基因组编写计划的主要目标是在10年内,在细胞系中实现大基因组的工程改造和检测,并且降低成本1 000倍以上。人类基因组编写计划(HGP-Write)还致力于为挑战人类健康的疾病提供解决方法,其潜在应用包括:培育可供移植的人体器官,细胞系全基因组重编码获得对病毒的免疫作用,将癌症抗性引入细胞系中用于癌症治疗,加速实现高产量、效益均衡的疫苗生产等。
5 结论与展望
目前,大尺度DNA分子组装仍然主要依赖微生物宿主细胞的同源重组系统在胞内进行。不同的宿主细胞的同源重组各有其特点,根据不同的组装需求选择合适的组装技术。基于常用宿主细胞大肠杆菌、枯草芽孢杆菌和酿酒酵母的体内重组机制发展了一系列体内组装技术,推动全基因组合成的研究。大肠杆菌中的噬菌体重组系统具有组装周期短、方便转移到其他表达宿主的优点,但是组装长度一般较小,很少用于全基因组合成。枯草芽孢杆菌重组系统组装量大,相对于传统的载体来说,BGM载体和iREX可操作性好,但是也存在着错误整合外源基因片段的风险,同时利用BGM作为载体也增加了大尺度DNA转移的难度。依赖于酿酒酵母高效的同源重组系统和便捷的分子操作手段,使之成为目前最受欢迎的大DNA组装技术。但是酵母组装也存在限制因素,比如DNA的GC含量、异源基因的细胞毒性和缺少原宿主的转录后修饰都可能影响大DNA组装效果。
随着合成基因组学的发展,对染色体规模的大DNA组装技术的开发将变得越来越重要,大DNA组装技术还需要在提升组装效率、降低组装成本、拓展组装能力和开发转移技术等方面不断发展。比如在相关机制深入揭示的基础上,开发新的分子生物学工具,突破更大、更复杂的大DNA组装技术,开发新的宿主用于构建含有特殊结构(例如具有高GC含量或高度重复序列)的大DNA。此外,开发通用型组装宿主,便于超大DNA向其他细胞体系转移也有迫切需要。
猜你喜欢
汉字汉语研究(2021年2期)2021-08-30
汉字汉语研究(2019年2期)2019-08-27
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
中国调味品(2017年2期)2017-03-20
中国调味品(2017年2期)2017-03-20
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
河北书画研究(2016年3期)2016-04-28
中国科技信息(2015年2期)2015-11-16
