
第三代EGFR抑制剂研究进展
2019-02-15 07:52:28孔月月饶国武
浙江化工 2019年1期
孔月月,饶国武
(浙江工业大学药学院,浙江 杭州 310014)
0 前言
EGFR是细胞膜表面的表皮生长因子受体,具有酪氨酸激酶(tyrosine kinase,TK)活性,EGFR在调控细胞的增殖、分化及凋亡占据重要的角色。通过抑制蛋白酪氨酸激酶的活性表达,可以有效抑制肿瘤的生长和增殖[1-2]。EGFR靶点成为近几年来研究的热点。迄今为止,美国食品药品管理局(FDA)分别在2003年和2004年批准了第一代EGFR靶向酪氨酸激酶抑制剂(TKIs)吉非替尼和厄洛替尼,第一代EGFR抑制剂的早期结果是令人振奋的,大多数具有活化突变的NSCLC患者早期治疗效果良好[3-4]。不幸的是,患者在几个月内产生了对第一代EGFR TKIs治疗的耐药性。获得性耐药性最常见的机制是外显子20中的次级T790M(苏氨酸替代甲硫氨酸)把控点突变[5]。
为了克服对第一代TKI获得性耐药性的问题,目前已经开发了几种第二代EGFR TKI。这些不可逆的EGFR TKI有可与EGFR的ATP结合裂隙中的Cys797的巯基形成共价键的迈克尔受体部分,由此增加抑制效力。第二代EGFR在治疗过程中逐渐出现了野生型的EGFR T790M靶点,进而对第二代TKI产生了耐药性[6-7]。
为了克服上述问题第三代EGFR抑制剂逐渐被开发出来,下面对近两年来已经上市(或临床)的第三代EGFR抑制剂和具有成药潜力的活性小分子进行总结。
1 已上市 (或临床)的第三代EGFR抑制剂
在临床治疗中靶点EGFR T790M产生了野生型突变和对第二代不可逆EGFR抑制剂的耐药,在临床用药上不得不加大用量达到药物作用的有效浓度,在加大药量的同时人体产生了严重的毒副反应,鉴于此针对耐药性和野生型EGFR T790M开发了高效低毒的第三代EGFR抑制剂。
1.1 Osimertinib
Osimertinib是由阿斯利康公司生产上市的药物,2015年11月,经优先审查后,美国FDA加速批准奥希替尼用于治疗转移性表皮生长因子受体 (EGFR)T790M突变阳性的非小细胞肺癌(NSCLC)。
Osimertinib是一种单苯胺基嘧啶化合物 (化合物1,Figure 1)。Osimertinib通过与EGFR激酶的ATP结合位点中的半胱氨酸-797残基形成不可逆共价键而产生作用。该化合物是不可逆的突变型选择性EGFR TKI(外显子19缺失EGFR IC50=12.92 nM,L858R/T790M EGFR IC50=11.44 nM,野生型EGFR IC50=493.8 nM)。它是目前唯一批准的适用于转移性EGFR T790M突变阳性非小细胞肺癌患者[8-9]的EGFR TKI。

Figure1 已上市(或临床)的第三代EGFR抑制剂
1.2 Olmutinib
Olmutinib(化合物 2,Figure 1)于 2016 年 5月获得韩国食品药品安全部(MFDS)批准[10-11],它最初是由Hanmi发现的,然后授权给勃林格殷格翰的开发和全球商业化权利。它用于治疗局部晚期或转移性表皮生长因子受体(EGFR)T790M突变阳性非小细胞肺癌[12]。
Olmutinib是一种不可逆激酶抑制剂,与突变型EGFR激酶结构域附近的半胱氨酸残基共价结合。HM61713在细胞系 H1975(L858R和T790M)和 HCC827(外显子 19缺失)[13-16]中引起有效抑制。HM61713对携带野生型EGFR的NSCLC细胞系 H358具有低效力(GI50为 2225 nM)[8-9]。在用H1975和HCC827异种移植模型的体内研究中,HM61713对肿瘤具有抑制生长作用而没有显示任何副作用[8-9]。
1.3 Nazartinib
Nazartinib(化合物 3, Figure 1)是由诺华公司研发的处于临床Ⅱ期的口服不可逆的第三代突变选择性表皮生长因子受体(EGFR)抑制剂[17]。
Nazartinib共价结合并抑制突变形式的EGFR(包括T790M EGFR突变体)的活性,从而阻止EGFR介导的信号转导[18]。EGF816优先抑制突变形式的EGFR,包括T790M(二次获得性耐药突变),与其他EGFR酪氨酸激酶抑制剂相比,可能对T790M介导的耐药肿瘤有治疗作用[19]。
1.4 Brigatinib
Brigatinib(化合物4,Figure 1)是一种口服药物,是选择性的双ALK/EGFR抑制剂。Brigatinib结合并抑制ALK激酶和ALK融合蛋白以及EGFR和突变形式[20],这将破坏它们的信号传导途径,从而抑制ALK激酶和EGFR激酶,并最终抑制易感肿瘤细胞中的肿瘤细胞生长[21-22]。Brigatinib已由美国Ariad(现该公司已被日本武田药品株式会社收购)提交,用于治疗对克唑替尼耐药的间变性淋巴瘤激酶(ALK)阳性局部晚期或转移性非小细胞肺癌(NSCLC)患者[23]。此药物在正处于Ⅰ/Ⅱ期临床试验阶段,因试验结果显示出较有前景的治疗效果,FDA授予其突破性治疗地位。该公司正在寻求FDA加速批准brigatinib,并计划要求对申请进行优先审查。
1.5 Naquotinib
Naquotinib(化合物5,Figure 1)是由Astellas公司开发的处于临床Ⅱ期的第三代不可逆EGFR小分子抑制剂。通过质谱发现Naquotinib通过与突变体EGFR(L858R/T790M)的激酶结构域中的半胱氨酸残基797共价结合,长效抑制EGFR磷酸化达24 h。在体外酶促和基于细胞的测定中,针对EGFR突变体(L858R,外显子19缺失,L858R/T790M和 del19/T790M)和 WT EGFR来评估Naquotinib。结果显示,在携带上述EGFR突变的 NSCLC细胞系中,Naquotinib对EGFR突变体的 IC50值为 8~33 nM,比对 WT EGFR抑制作用更强(IC50值为230 nM)[24]。值得关注的是Naquotinib对于在用其他EGFR TKIs包括AZD9291和CO1686用药中出现耐药的患者具有作用效果。
1.6 其他第三代EGFR抑制剂
除了上述介绍的几种小分子药物,还有一些在临床试验中被终止的小分子药物。Rociletinib(化合物6,Figure 1)又名CO1686,由阿维拉治疗学和发起人和克洛维斯肿瘤学联合开发,在2016年4月临床Ⅱ/Ⅲ期临床试验阶段被美国FDA驳回。WZ4002(化合物 7,Figure 1)是具有突破性进展的第三代EGFR小分子抑制剂。但由于化合物的专利归属问题使其在临床研究中停滞。
2 具有成药潜力的第三代EGFR抑制剂
2016年,Hao等人[25]基于先前报道的不可逆EGFR抑制剂(化合物8,Figure 2)并根据其结构的设计,结合分析其结合模型并比较T790M突变体和野生型 (WT)EGFR激酶之间的关守口袋的差异来对化合物8进行改造。在这些结果的指导下,发现了一种新型的6,7-二氧代-6,7-二氢蝶啶骨架,并且在N5-位进行疏水修饰以增强非极性接触并提高突变选择性和对WT(EGFR)的抑制作用。最后,确定了最具代表性的化合物9(化合物 9,Figure 2)。 其合成如图(Scheme 1)。 化合物9对H1975癌细胞表现出强烈的抗增殖活性,而对A431癌细胞的作用不明显。化合物9的进一步体内抗肿瘤效力评估表明,通过以50 mg/kg-1d-1剂量对H1975NSCLC异种移植小鼠口服给药14天,其结果显示化合物9能明显抑制小鼠模型中的肿瘤生长。由于化合物9具有良好的选择性和体内抗肿瘤作用,它极有可能被用作进一步发展为一种可以克服EGFR耐药突变的先导化合物。
2017年,Song等人[26]使用由具有吗啉官能团的结构修饰的二苯基嘧啶衍生物(Mor-DPPYs)组成的潜在的新EGFR T790M抑制剂来改善吉非替尼抗性的非小细胞肺癌(NSCLC)治疗的活性和选择性。最有希望的抑制剂化合物10(化合物10,Figure 2),不仅显示出高活性EGFR T790M/L858R激酶(IC50=0.71 nM),而且还能够抑制携带EGFRT790M突变的H1975细胞的复制。其合成如图(Scheme 2)。总的来说,这一发现为进一步开发抗NSCLC药物提供了新的有前景的先导化合物。

Figure2 具有成药潜力的第三代EGFR抑制剂
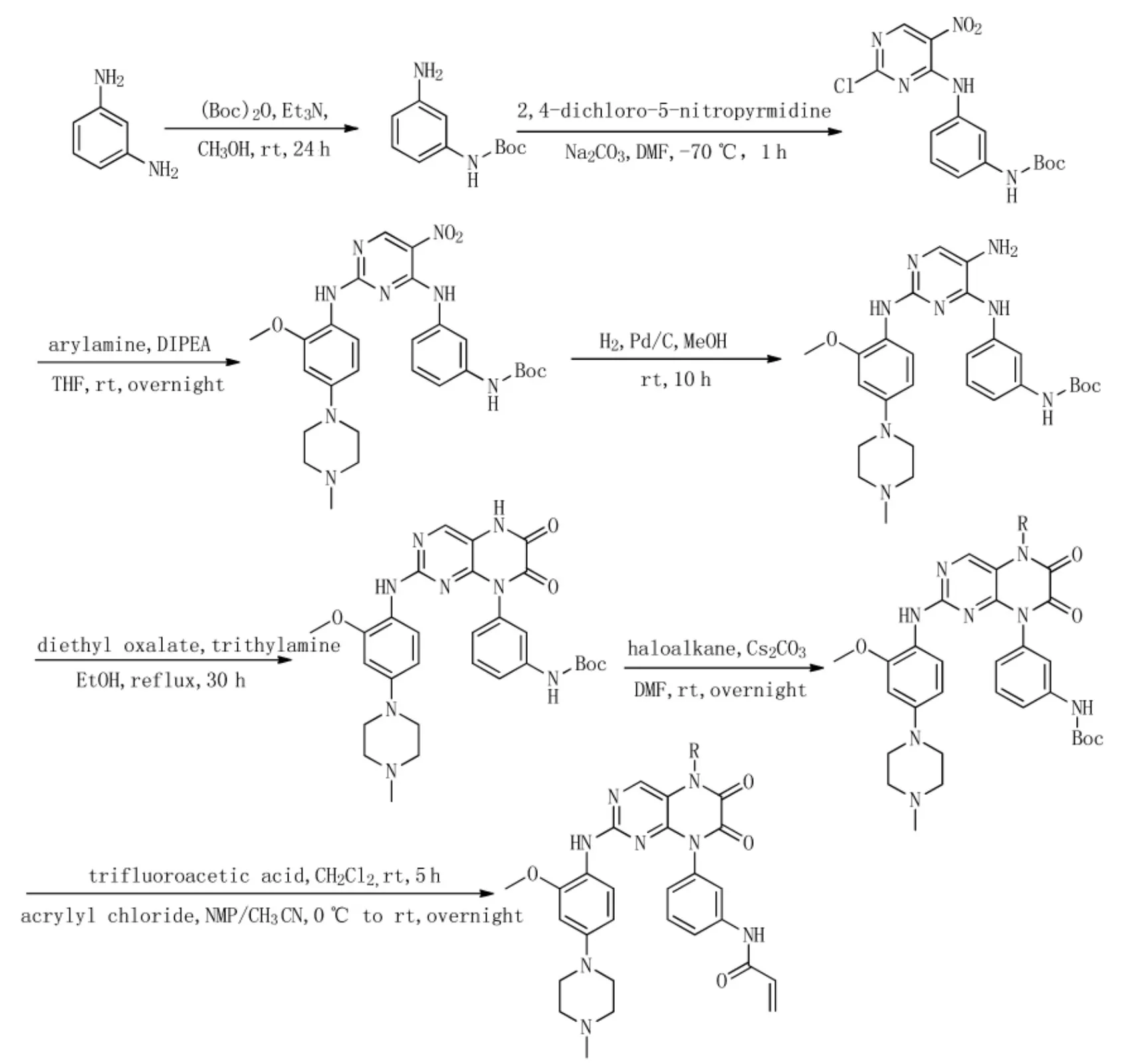
Scheme1

Scheme2

2017年,Tomassi等人[27]通过分子模拟分析化合物与蛋白质空腔的结合模式设计了独特氨基吡唑类化合物。并获得了15个能有效抑制EGFR耐药突变变体同时对野生型有选择性的衍生物。其中化合物11(化合物11,Figure 2)显示对H1975细胞存活力的有效抑制,EC50为694 nM,并且对HCC827细胞也有活性EC50为459 nM。此外,通过蛋白质印迹分析这些抑制剂也显示出抑制激酶磷酸化的作用。其合成如图(Scheme 3)。总之,新型的选择性吲唑基耐药EGFRL858R/T790M抑制剂被开发出来,并显示出强的抑制作用,它极有可能被用作进一步发展为一种可以克服EGFR耐药突变的先导化合物。
2017 年,Yu 等人[28]进行吡啶并[2,3-d]嘧啶-7-酮的结构优化,合成一系列具有改善的药代动力学性质的新的选择性EGFR T790M抑制剂。其中最有希望的是化合物12(化合物12,Figure 2),能够有效地抑制EGFR L858R/T790M激酶并抑制H1975细胞的增殖,其IC50值分别为2.0 nM和40 nM。化合物12剂量依赖性地诱导细胞减少EGFR的磷酸化和NCI-H1975细胞中ERK的下游活化。并且口服生物利用度值为16%。其合成如图(Scheme 4)化合物12可以发展为有希望的先导化合物,以克服获得的非小细胞肺癌(NSCLC)患者的抗性。
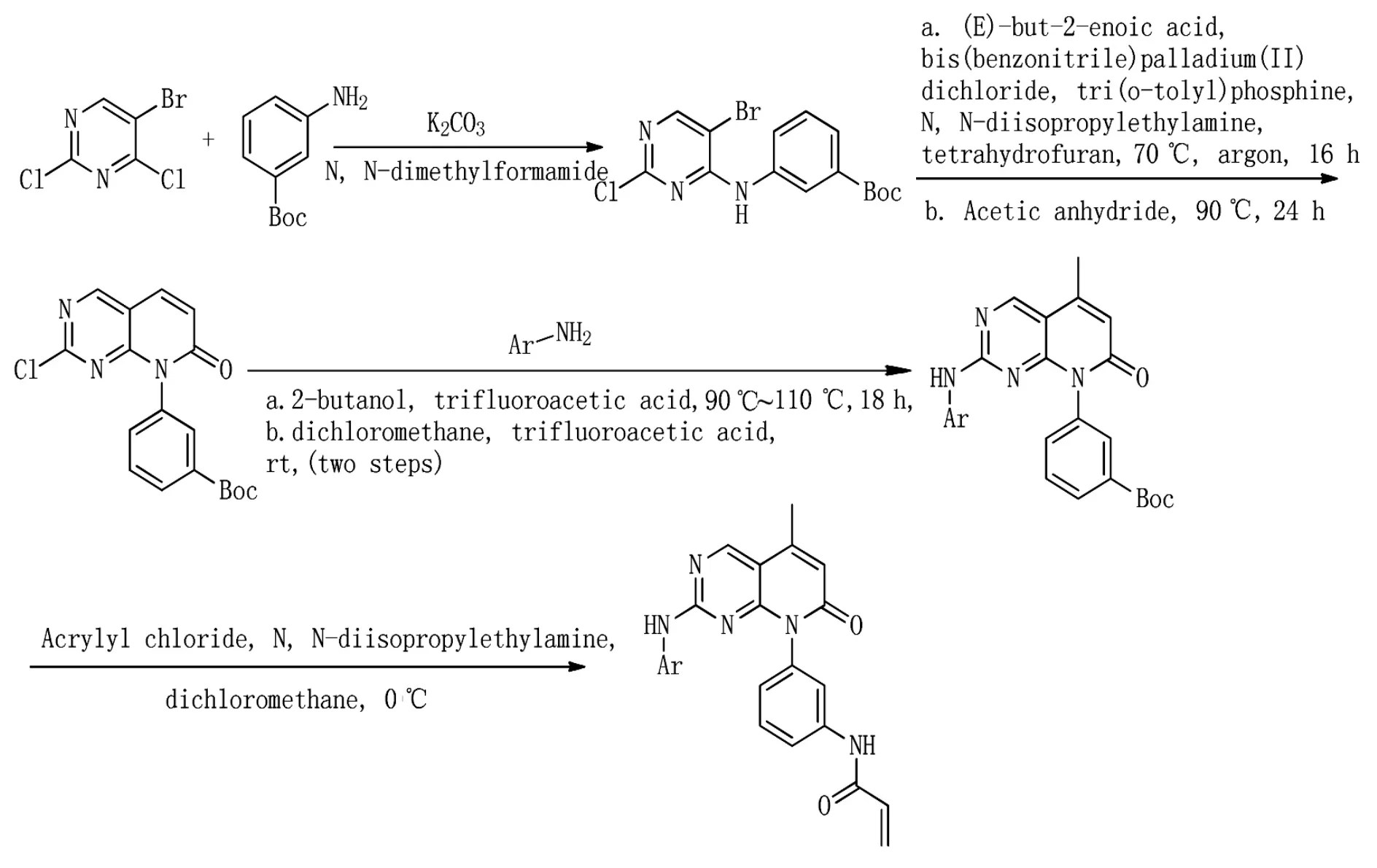
Scheme4
2017年,Zhang等人[29]基于市售AZD9291与EGFR T790M的建模结合模式,设计并合成了一系列 5,6-二氢-4H-吡咯并 [3,2,1-ij] 喹啉衍生物,目的是克服T790M/L858R双重突变引起的耐药性。最有效的化合物13(化合物13,Figure 2)对于单L858R突变和双重T790M/L858R突变型EGFR显示出优异的酶抑制活性和选择性,IC50值亚纳摩尔级,并且对野生型EGFR具有超过8倍的选择性。化合物13表现出良好的微粒体稳定性和药代动力学性质,并且对hERG离子通道的结合亲和力低于AZD9291,并且对携带T790M/L858R的非小细胞肺癌 (NSCLC)细胞H1975显示出强抗增殖活性,在人NSCLC(H1975)异种移植小鼠模型中表现出体内抗癌效力。其合成如图(Scheme 5)。
3 总结
自第一代EGFR抑制剂吉非替尼和厄洛替尼2002年获准用于治疗NSCLC疾病,2004年分别推出了一些更有效的抗EGFR药物。然而,这些药物在约50%的临床治疗患者中产生了抵抗,单一T790M点突变在EGFR在第一代重复治疗后出现耐药,使这些药物失去重大有效治疗NSCLC疾病。因此,寻找对突变EGFR T790M特别有效的药物仍然很紧急。最近,大量的小分子,包括新型临床候选药物罗西替尼,osimertinib和HM81713,已被发现强力抑制EGFR T790M突变激酶。特别是罗西替尼和osimertinib,代表了领先的临床开发中的第三代不可逆EGFR抑制剂。希望它们的批准将为患者的治疗提供重大突破EGFR T790M突变的NSCLC。更令人兴奋的是,持续专注于这一领域的努力也产生了一些甚至更多的选择性抑制剂。然而在寻找更加有效,更加有选择性的抑制剂还有很长的道路要走。
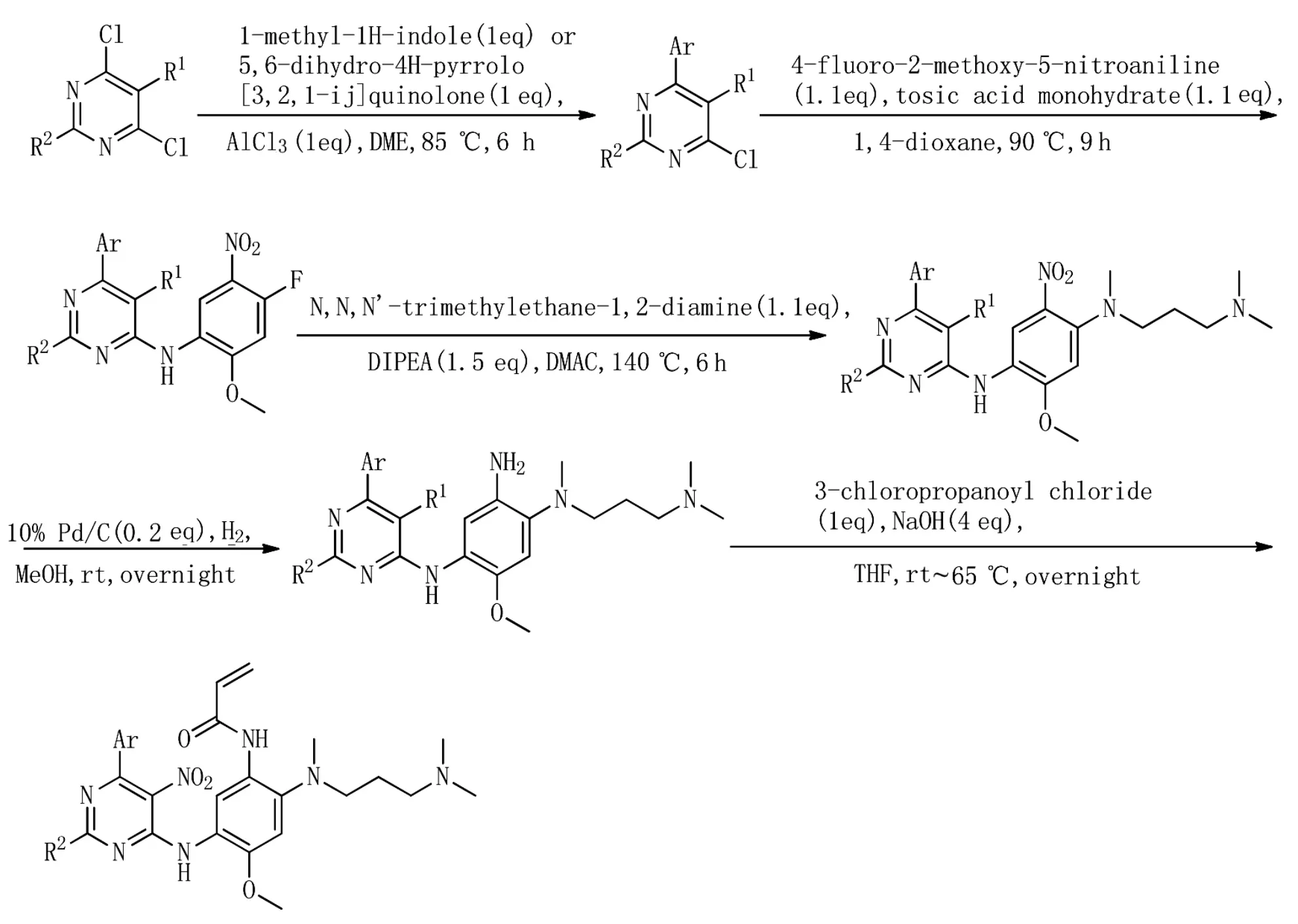
Scheme5
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
第一财经(2019年8期)2019-08-26 17:53:46
上海农业学报(2017年3期)2017-04-10 12:39:26
中国医药生物技术(2015年4期)2015-12-26 08:26:36
哈尔滨医药(2015年2期)2015-12-01 03:57:13
学习月刊(2015年14期)2015-07-09 03:37:48
中国当代医药(2015年16期)2015-03-01 02:03:13
物理化学学报(2015年5期)2015-02-28 17:34:47
中国药理学通报(2014年2期)2014-05-09 08:22:39
