
超高压脲包法提高模拟亚麻籽油中多不饱和脂肪酸分离效率
2019-01-28 06:09朱凯莉陈婧超范清苹惠爱玲
食品科学 2019年1期
朱凯莉,陈婧超,范清苹,惠爱玲*
(合肥工业大学食品科学与工程学院,安徽 合肥 230009)
亚麻籽油含丰富的不饱和脂肪酸,其中以亚油酸和亚麻酸为主的多不饱和脂肪酸(poly unsaturated fatty acids,PUFA)质量分数高达60%以上[1]。亚油酸和亚麻酸是人体必需脂肪酸,其在治疗皮炎、风湿性关节炎、心血管疾病、高血压、哮喘、过敏症和抑制肿瘤生长方面具有一定功效[2-5]。因此,对亚麻籽油中亚油酸和亚麻酸的分离纯化或富集研究尤为重要。尿素包合法(脲包法)是分离纯化亚油酸、亚麻酸等PUFA的常用方法[6-7]。脲包法对设备要求简单,且易操作,但存在包合时间长[8-10]、产品得率低[11-12]、单不饱和脂肪酸与PUFA选择性差[13]等缺点。为此,一些研究者对传统脲包(低温脲包、冷冻脲包)过程加以改进,如采用多次包合[14-15]、缓慢降温[16]、离心[17]、溶剂结晶法和脲包法联用[18]、脲包法与选择性加氢结合[19]等方法提高PUFA的纯度;通过响应面优化试验[20]、搅拌梯度冷却结晶[21]以缩短包合时间;采用快速降温(1 min内将温度从室温降到-60 ℃)使游离脂肪酸的分离更具有选择性[22-24];采用超声波处理以提高重质油中链烷烃和环烷烃分离效果[25]。以上改进措施在某种程度上提高了传统脲包法的分离效率,但产品得率仍然偏低。
Brown等[26]研究发现施加一定压力可使2,10-十一烷二酮-尿素包合物结构发生一些不可逆的变化。高压处理对饱和脂肪酸的影响较不饱和脂肪酸更为显著[27]。袁野等[28]采用低温协同超高压脲包法分离大豆油脱臭馏出物中饱和脂肪酸乙酯工艺,当脲包混合液在4 ℃下预冷90 min并用300 MPa超高压处理60 min时,非包合相(滤液)中饱和脂肪酸乙酯质量分数由15.22%减少至2.83%,不饱和脂肪酸乙酯质量分数由37.65%提高至60.01%。以上研究结果提示,将高压技术引入传统脲包过程,有望取得与冷冻脲包法类似的分离效果,并可能大幅缩短包合周期。
本研究以模拟亚麻籽油混合脂肪酸(fatty acid mixed,MFA)为研究对象,采用超高压脲包法富集PUFA,优化包合工艺参数以提高PUFA的分离效率;采用差示扫描量热(differential scanning calorimetry,DSC)仪和扫描电子显微镜(scanning electron microscope,SEM)研究压力对尿素包合物晶体热力学性质与晶体形貌的影响,初步解析超高压处理提高PUFA分离效率的机理,为超高压技术应用于油脂PUFA分离提供理论依据和技术支持。
1 材料与方法
1.1 材料与试剂
棕榈酸、硬脂酸、油酸、亚油酸、亚麻酸 合肥博美生物科技有限责任公司;棕榈酸甲酯(99.0%)、硬脂酸甲酯(98.5%)、油酸甲酯(99.0%)、亚油酸甲酯(99.4%)、亚麻酸甲酯(98.0%) 德国Dr.Ehrenstorfer公司;十七烷酸甲酯 美国Uu-chek公司;尿素、盐酸、体积分数95%乙醇溶液、无水甲醇、浓硫酸、正己烷 国药集团化学试剂有限公司。
1.2 仪器与设备
YCB630/2.5型食品超高压处理装置 中国兵器工业第五二研究所;Claus 600气相色谱仪(gas chromatography,GC) 珀金埃尔默仪器(上海)有限公司;Q200 DSC仪 美国TA公司;SU8020场发射SEM日本日立公司;X’Pert Pro MPD X射线衍射(X-ray diffraction,XRD)仪 荷兰帕纳科公司。
1.3 方法
1.3.1 亚麻籽油MFA制备
模拟亚麻籽油中MFA体系,将5 种主要脂肪酸(棕榈酸、硬脂酸、油酸、亚油酸、亚麻酸,其中亚油酸、亚麻酸为PUFA)按一定比例混合均匀。GC定量检测显示其质量比为7∶5∶19∶15∶54。
1.3.2 脲包法富集亚麻籽油中PUFA
低温脲包法流程为:尿素、95%(体积分数,下同)乙醇溶液、MFA混合→85 ℃加热回流→搅拌30 min→于4 ℃放置18 h→待抽滤处理。
冷冻脲包法流程为:尿素、95%乙醇溶液、MFA混合→85 ℃加热回流→搅拌30 min→于-18 ℃放置18 h→待抽滤处理。
超高压脲包法流程为:尿素、95%乙醇溶液、MFA混合→85 ℃加热回流→搅拌30 min→于聚乙烯袋中冷却30 min→待高压处理。
预实验发现超高压处理之前的预冷却温度及超高压处理的保压时间对PUFA分离效果影响较小,从使包合物充分结晶且经济实用的角度考虑,选择高压处理之前在20 ℃下冷却30 min,保压20 min。因此,本研究仅考察不同尿素/MFA质量比及压力大小对非包合相(滤液)中PUFA富集效果及包合相中PUFA非选择性包合的影响。
尿素/MFA质量比的影响:固定MFA2 g、95%乙醇溶液14 mL,尿素用量分别为1、2、3、4 g,脲包混合液在20 ℃下冷却30 min,之后在300 MPa下保压20 min使其结晶。
压力大小的影响:固定MFA2 g、95%乙醇溶液14 mL、尿素3 g,脲包混合液在高压前于20 ℃冷却30 min,其后分别采用0.1、100、200、300、400 MPa压力保压20 min使其结晶。
1.3.3 包合相及非包合相中PUFA得率的测定
对脲包法得到的包合物立即进行抽滤,分别得到包合相(晶体)及非包合相(滤液)。将包合相晶体晾干,取1 g置于10 mL具塞试管中,加入3 mL体积分数15%盐酸溶液置于50 ℃水浴至晶体完全溶解[29],用正己烷萃取脂肪酸,减压浓缩得包合相脂肪酸。将非包合相中滤液减压浓缩除去乙醇,加入正己烷萃取脂肪酸,水洗去除尿素,无水硫酸钠干燥,减压浓缩得非包合相脂肪酸。将上述脂肪酸分别甲酯化后用GC定量检测其中PUFA质量分数,并按下式计算非包合相PUFA得率(Yf)。

式中:mf表示非包合相中M F A的质量/g;ωf表示非包合相中P U F A的质量分数/%;m表示原料中M F A的质量/g;ω表示原料中PUFA的质量分数/%。
脂肪酸甲酯化:取脂肪酸200 mg,加入1 mL甲醇和0.1 mL浓硫酸,混匀后65 ℃水浴加热1 h,加入1 mL正己烷,离心,保留上清液待用。取上清液20 µL,加入200 µL含3 mg/mL十七烷酸甲酯(内标物)的正己烷溶液,并用正己烷定容至0.5 mL,过0.22 µm有机滤膜,待GC检测。
GC检测条件:PE Elite-WAX ETR 毛细管柱(30 m×0.25 mm,0.25 μm);氢火焰离子化检测器,检测器温度300℃;空气流速400 mL/min,氢气流速40 mL/min;进样口温度250 ℃;载气流速1 mL/min;分流比12∶1;进样量0.5 μL;升温程序:200 ℃保持10 min。
1.3.4 PUFA质量分数的GC定量检测
方法特异性:对加入内标物前后的MFA进行GC分析,具体方法如下。以正己烷为溶剂,配制质量浓度为3 mg/mL的十七烷酸甲酯溶液。取两份200 mg MFA甲酯化并稀释后的上清液,其中一份加入200 µL十七烷酸甲酯溶液,另一份加入等体积的正己烷,利用GC检测。
标准曲线:分别取一定质量的棕榈酸甲酯、硬脂酸甲酯、油酸甲酯、亚油酸甲酯、亚麻酸甲酯标准品,用正己烷混合溶解,每种脂肪酸甲酯的质量浓度分别为6.0、4.0、9.6、8.6、10.0 mg/mL。用正己烷稀释成7 个质量浓度梯度的MFA甲酯标准品溶液,内标物质量浓度均为1.2 mg/mL,GC检测,以每种脂肪酸甲酯标准品与内标物的峰面积比(y)对每种脂肪酸甲酯溶液质量浓度(x/(mg/mL))进行线性回归分析,求得5 种脂肪酸甲酯在相应质量浓度范围内的标准曲线,其相关系数(R)应大于0.99。
精密度实验[30]:用正己烷将MFA甲酯标准品溶液稀释至棕榈酸甲酯、硬脂酸甲酯、油酸甲酯、亚油酸甲酯、亚麻酸甲酯质量浓度分别为1.20、0.80、1.92、1.72、2.00 mg/mL,其中内标物质量浓度为1.2 mg/mL,用GC对其重复测定6 次,比较测定结果。
回收率实验[31]:本研究采用加标回收法测定回收率,取已知质量浓度的MFA甲酯样品,加入5 种脂肪酸甲酯标准品,使棕榈酸甲酯、硬脂酸甲酯、油酸甲酯、亚油酸甲酯、亚麻酸甲酯标准品质量浓度相对于已知质量浓度分别增加0.5、0.5、1.5、1.5、4.0 mg/mL,每种样品取6 份作为平行样,采用GC内标法定量检测,通过每种标准品的加入量和检出量评价回收率。
稳定性实验[32]:取处理后的MFA样品,24 h内每隔2 h取待测液进行GC检测。
1.3.5 包合相晶体分析
DSC分析:准确称取3 mg包合物晶体放入铝制坩埚中,密闭放入样品池中,并用空白坩埚作对照。设置程序升温样品初始温度80 ℃,以10 ℃/min的速率升温至160 ℃。场发射SEM观察:参考鉏晓艳等[33]的方法。XRD分析:参考陈迪钊等[34]的方法并略作改动,扫描范围为7°~40°。
1.4 数据统计分析
2 结果与分析
2.1 PUFA质量分数的GC定量分析
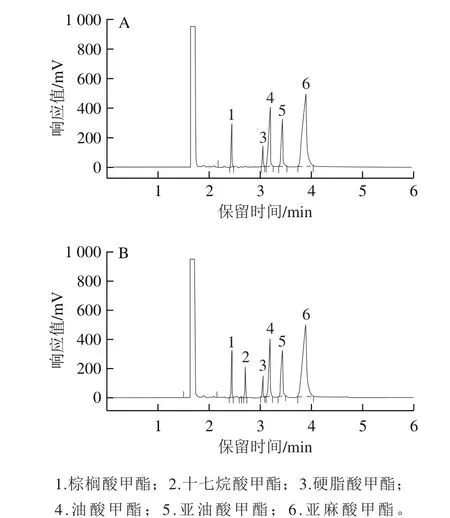
图1 MFA甲酯未加(A)/加(B)内标物的比较Fig.1 Chromatograms of MFA methyl esters without (A)/with (B)internal standard substance
图1 A是模拟亚麻籽油中5 种主要脂肪酸的GC图谱,图1B是加入内标物十七烷酸甲酯后的图谱。可以看出十七烷酸甲酯与被检测脂肪酸有明显的分离,且其保留时间处于5 种脂肪酸中间,适于作为脂肪酸质量分数定量检测的理想内标。
棕榈酸甲酯、硬脂酸甲酯、油酸甲酯、亚油酸甲酯、亚麻酸甲酯在0.12~6.00、0.08~4.00、0.19~9.60、0.17~8.60、0.20~10.00 mg/mL质量浓度范围内的线性回归方程分别为y=0.705 0x+0.029 8、y=0.774 3x+0.020 8、y=0.817 1x+0.053 5、y=0.806 5x+0.033 1、y=0.779 2x+0.050 7,R在0.997 6~0.998 5之间,线性关系良好,说明该实验方法可用于亚麻籽油中5 种脂肪酸质量浓度的准确测定。精密度实验结果得出5 种脂肪酸甲酯的相对标准偏差(relative standard deviation,RSD)均小于2%,表明本方法可靠、准确,具有重复性;回收率实验中5 种脂肪酸定量分析的回收率在94.44%~103.28%之间,符合实验要求,被测脂肪酸的RSD均小于3%,重复性较好;稳定性实验结果中5 种脂肪酸甲酯的质量浓度RSD均小于4%,表明脂肪酸甲酯样品在24 h内比较稳定。
2.2 超高压脲包法分离富集PUFA分析
2.2.1 尿素/MFA质量比分析
在传统脲包法分离过程中,尿素/脂肪酸质量比决定了尿素-脂肪酸包合物的形成,并最终影响饱和脂肪酸的包合效果[35]。在超高压脲包法分离中,随着尿素/MFA质量比的增加,非包合相中PUFA质量分数逐渐增加。如图2所示,当尿素/MFA质量比达4∶2时,非包合相中PUFA质量分数由MFA中的69%增加至99.16%。与此同时,包合相中也检测到50.59%的PUFA,即大于50%的PUFA被非选择性包合,这一结果也可从非包合相中PUFA得率仅为36.62%得以印证。而当尿素/MFA质量比为3∶2时,尽管非包合相中PUFA质量分数(96.05%)与质量比4∶2(99.16%)时相比略有降低,但非包合相中PUFA得率高达81.81%,这得益于PUFA在尿素/MFA质量比为3∶2时被非选择性包合的损失(37.87%)与质量比4∶2(50.59%)时相比有所降低。综合考虑PUFA质量分数及得率,选择尿素/MFA质量比为3∶2且尿素-MFA与95%乙醇溶液料液比为5∶14进行后续实验。

图2 尿素/MFA质量比对PUFA分离效果的影响Fig.2 Effect of urea/MFA ratio on separation eff i ciency of PUFA
2.2.2 超高压处理压力分析

图3 处理压力对PUFA分离效果的影响Fig.3 Effect of pressure on separation eff i ciency of PUFA
一般认为:脂肪酸与尿素包合过程是一个反应结晶(沉淀)过程,超高压处理有助于促进尿素-脂肪酸包合结晶过程[21]。如图3所示,处理压力对包合相及非包合相中PUFA质量分数影响较小,然而施加压力明显促进了非包合相中PUFA得率的变化。当处理压力小于300 MPa时,非包合相中PUFA得率随着压力的增大而逐步增加;而当压力达到400 MPa时,非包合相中PUFA得率(61.57%)较300 MPa(81.81%)时降低了24.74%(P<0.05)。故超高压脲包法处理压力以300 MPa为宜。
2.3 超高压脲包法与冷冻脲包法的对比
2.2 节结果确立了超高压脲包法富集亚麻籽油中PUFA的较优工艺:尿素/MFA质量比为3∶2且尿素-MFA与95%乙醇溶液料液比为5∶14,20 ℃冷却后对脲包混合液施加300 MPa压力,保压20 min。以此料液比为基础,同时比较4 ℃低温脲包、-18 ℃冷冻脲包对PUFA富集效果影响。由图4可知,超高压脲包法所得非包合相中PUFA质量分数(96.05%)与4 ℃(95.68%)或-18 ℃(97.41%)包合18 h的效果类似,但其非包合相中PUFA得率分别提高了28.92%和41.39%,这也说明超高压脲包法非包合相中PUFA得率的提高不止是由非包合相中PUFA质量分数下降导致,压力在一定程度上促进了非包合相中PUFA得率的提高,且使包合时间大幅缩短,超高压脲包法大幅提高了PUFA的分离效率。

图1 不同结晶条件对PUFA分离效果的影响Fig.1 Effect of crystallization conditions on separation eff i ciency of PUFA

图5 不同结晶条件下的尿素包合物晶体XRD图谱Fig.5 XRD of urea inclusion complex crystals under different crystallization conditions
由于超高压脲包法可达到冷冻脲包法相似的分离效果,因此将二者的包合物作晶体XRD分析,结果如图5所示。尿素包合物晶体衍射峰对应六方晶系晶体结构[36]。冷冻与超高压条件下出现强烈衍射峰时所在的角度相近,由Jade软件得出冷冻条件下晶面间距分别在7.160 7、4.149 7、3.895 8、3.585 2、3.408 6、2.704 0、2.6287 Å时有强烈的衍射峰,超高压条件下晶面间距分别在7.129 4、4.118 7、3.859 5、3.554 0、3.394 8、2.693 3、2.6206 Å时有强烈的衍射峰。两种条件下晶体出峰的晶面间距无显著性差异(P>0.05)。
2.4 压力对尿素包合物晶体性质的影响
2.4.1 DSC分析结果
为探究超高压脲包过程中非包合相中PUFA得率随压力变化的原因,用DSC仪测试了不同压力条件下尿素包合物晶体热力学性质。尿素包合物晶体熔化是吸热的过程,主要分为两个熔化阶段,第1阶段是尿素包合物六方晶系晶体的熔化阶段,第2阶段是四方晶系尿素晶体的熔化阶段[36]。在有机溶剂中,尿素包合脂肪酸时会形成六棱柱框架[37],尿素-油酸包合物晶体属于六方晶系,其晶体熔点为117.34 ℃[36]。图6中DSC曲线的第1个阶段峰温均接近于六方晶系晶体的熔点,故选取不同压力条件下的DSC曲线中第1个峰进行分析,结果如表1所示。

图6 不同处理压力尿素包合物DSC曲线Fig.6 DSC curves of urea inclusion complexes at different pressures
由表1可知,高压包合晶体样品ΔT明显高于未经超高压处理(0.1 MPa)样品。经过超高压处理后,其晶体熔融温度范围较未经高压处理时更宽、峰温更高,说明超高压处理后晶体平均厚度较厚,且压力从0.1 MPa上升至300 MPa时,晶体峰温逐渐升高,说明晶体厚度随压力的升高而增加;400 MPa处理时峰温降低,可能是由于压力过大导致晶体六棱柱结构在某种程度上被破坏。超高压处理包和物晶体ΔH较未经超高压处理的样品高,其中经300 MPa超高压处理后样品ΔH较未经超高压处理样品高36.68%,可知超高压处理所得晶体中,其六方晶系的晶体熔融时吸热较多,说明超高压处理可在一定程度上促进尿素包合物结晶生成六方晶系晶体,从而促使非包合相中PUFA得率提高。

表1 不同处理压力尿素包合物DSC曲线分析Table1 Thermodynamic parameters from DSC curves of ureainclusion complexes at different pressures
2.4.2 SEM观察结果

图7 不同处理压力尿素包合物的SEM图Fig.7 SEM images of urea inclusion complexes at different pressures
由图7可以看出,未经超高压处理时样品的晶体形态粗细不均、长短不一,有些晶体并未形成六棱柱状态,说明尿素并未完全包合脂肪酸,使得非包合相中PUFA质量分数较超高压处理时低;而且其晶体分布较为松散,这可能是由于晶体生长环境不固定,导致其在抽滤过程中非包合相液体易残存于晶体中,最终使得非包合相中PUFA得率较低。当施加100 MPa压力时,尿素包合物晶体形态规则,呈长针柱型,能看出明显的六棱柱晶体形貌,晶体大小分布相对0.1 MPa处理时较为均匀,说明在此条件下尿素可以相对成功地包合脂肪酸,使得非包合相中的PUFA质量分数有所增高,但晶体分布仍较为松散。当压力增加到300 MPa时,晶体呈短针状柱体,尿素优先选择性包合结构简单的饱和脂肪酸,使得PUFA较难进入尿素包合物框架,且在300 MPa处理时,晶体分布明显比0.1、100 MPa处理时密集,在抽滤时非包合相液体不易残存于晶体中,从而使得300 MPa处理时非包合相中PUFA得率最高。在400 MPa处理时,尿素包合物晶体呈六棱柱型,但可以看出有较多是断裂后短小的六棱柱晶体,这可能是由于压力过高导致包合条件较剧烈,从而使已经结合的尿素分子能量过高,当尿素晶体达到挣脱结晶体所需能量后就会从结晶体上脱离,原来成功包合脂肪酸的晶体结构被破坏,已包合的饱和脂肪酸被释放出来,使得非包合相中PUFA得率下降。
通过对不同压力下包合相晶体的热力学性质和形貌比较,初步说明了超高压作用有助于促进包合物晶体稳定,也初步解析了高压脲包法较传统脲包法PUFA得率提高的原因。
3 结 论
超高压脲包法富集模拟亚麻籽油PUFA较优工艺为:尿素/MFA质量比为3∶2且尿素-MFA与95%乙醇溶液料液比为5∶14,脲包混合液在20 ℃下冷却30 min后,采用300 MPa超高压处理20 min,此时非包合相中PUFA质量分数为96.05%,得率为81.81%。对于模拟亚麻籽油MFA中PUFA的富集,超高压脲包法所得非包合相中PUFA质量分数(96.05%)与低温(95.68%)或冷冻(97.41%)脲包法分离效果类似,但得率相应分别提高28.92%和41.39%。超高压脲包法富集亚麻籽油PUFA,其尿素包合物的晶面间距与冷冻脲包法无显著性差异。当处理压力小于300 MPa时,随着压力升高,晶体的热力学性质趋于稳定,六方晶系晶体熔融所需能量增加,且晶体形态更规则,晶体分布更密集,这也解释了高压脲包法提高PUFA分离效率的原因。
猜你喜欢
林业科技(2020年3期)2021-01-21
山东化工(2020年9期)2020-06-01
中成药(2018年8期)2018-08-29
中成药(2018年5期)2018-06-06
中成药(2018年5期)2018-06-06
天然产物研究与开发(2018年3期)2018-05-07
中成药(2017年12期)2018-01-19
中成药(2017年8期)2017-11-22
中成药(2017年5期)2017-06-13
中国粮油学报(2016年5期)2016-01-23
